

Transformation Asymmetry and the Evolution of the Bacterial Accessory Genome
- PMID: 29211859
- PMCID: PMC5850275
- DOI: 10.1093/molbev/msx309
Transformation Asymmetry and the Evolution of the Bacterial Accessory Genome
Abstract
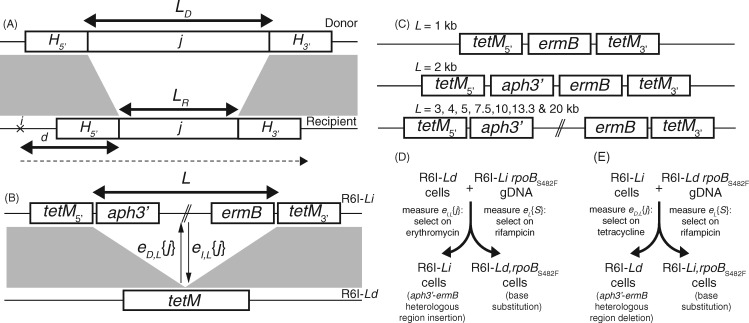
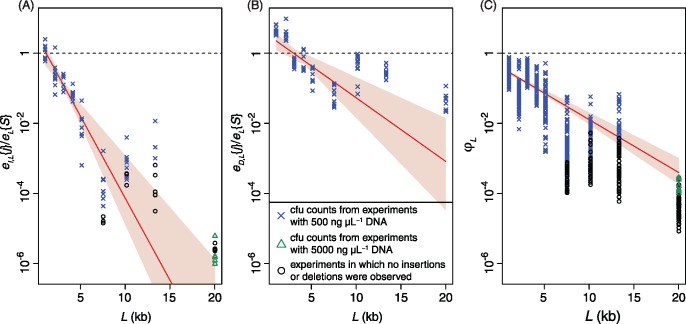
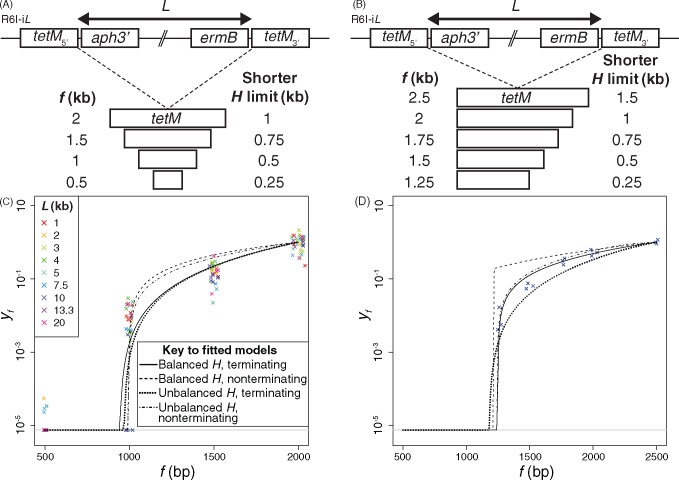
Bacterial transformation can insert or delete genomic islands (GIs), depending on the donor and recipient genotypes, if an homologous recombination spans the GI's integration site and includes sufficiently long flanking homologous arms. Combining mathematical models of recombination with experiments using pneumococci found GI insertion rates declined geometrically with the GI's size. The decrease in acquisition frequency with length (1.08×10-3 bp-1) was higher than a previous estimate of the analogous rate at which core genome recombinations terminated. Although most efficient for shorter GIs, transformation-mediated deletion frequencies did not vary consistently with GI length, with removal of 10-kb GIs ∼50% as efficient as acquisition of base substitutions. Fragments of 2 kb, typical of transformation event sizes, could drive all these deletions independent of island length. The strong asymmetry of transformation, and its capacity to efficiently remove GIs, suggests nonmobile accessory loci will decline in frequency without preservation by selection.
Keywords: bacterial evolution; horizontal gene transfer; mobile elements; pneumococcus; recombination; transformation.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Adams A. 1972. Transformation and transduction of a large deletion mutation in Bacillus subtilis. Mol Gen Genet. 118(4):311–322. - PubMed
-
- Baumdicker F, Hess WR, Pfaffelhuber P.. 2012. The infinitely many genes model for the distributed genome of bacteria. Genome Biol Evol. 4(4):443–456.http://dx.doi.org/10.1093/gbe/evs016 - DOI - PMC - PubMed
-
- Claverys JP, Lefevre JC, Sicard AM.. 1980. Transformation of Streptococcus pneumoniae with S. pneumoniae-lambda phage hybrid DNA: induction of deletions. Proc Natl Acad Sci U S A. 77(6):3534–3538.http://dx.doi.org/10.1073/pnas.77.6.3534 - DOI - PMC - PubMed
-
- Collins RE, Higgs PG.. 2012. Testing the infinitely many genes model for the evolution of the bacterial core genome and pangenome. Mol Biol Evol. 29(11):3413–3425.http://dx.doi.org/10.1093/molbev/mss163 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
