

Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy
- PMID: 29212899
- PMCID: PMC5802346
- DOI: 10.1161/CIRCGENETICS.117.001780
Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy
Abstract
Background: Restrictive cardiomyopathy (RCM) is a rare cardiomyopathy characterized by impaired diastolic ventricular function resulting in a poor clinical prognosis. Rarely, heritable forms of RCM have been reported, and mutations underlying RCM have been identified in genes that govern the contractile function of the cardiomyocytes.
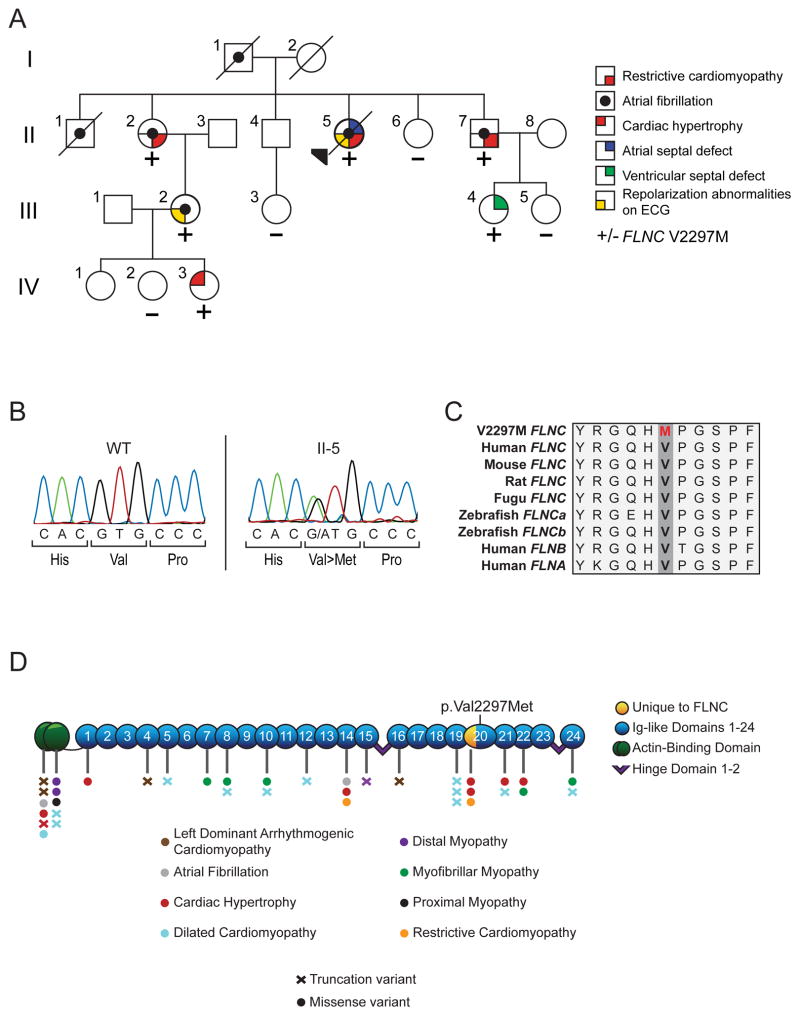
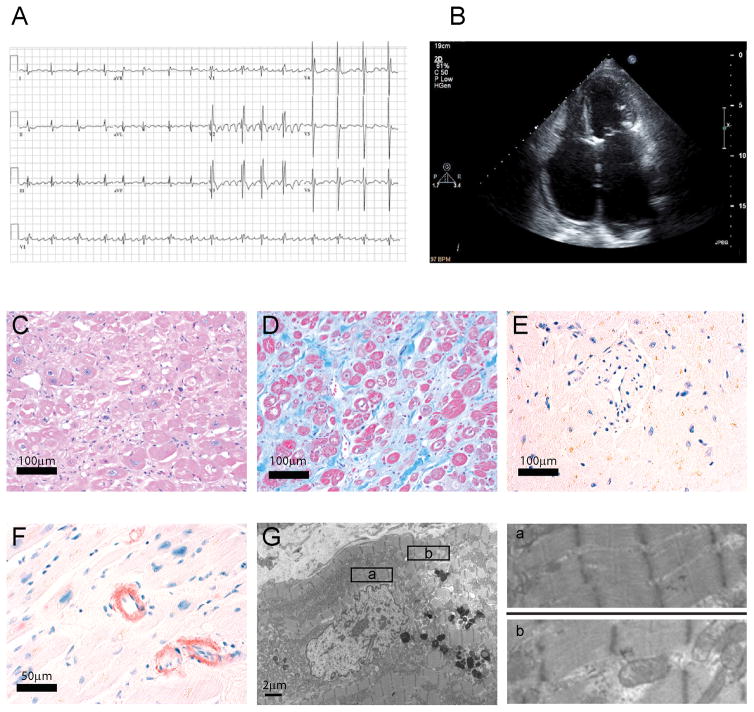
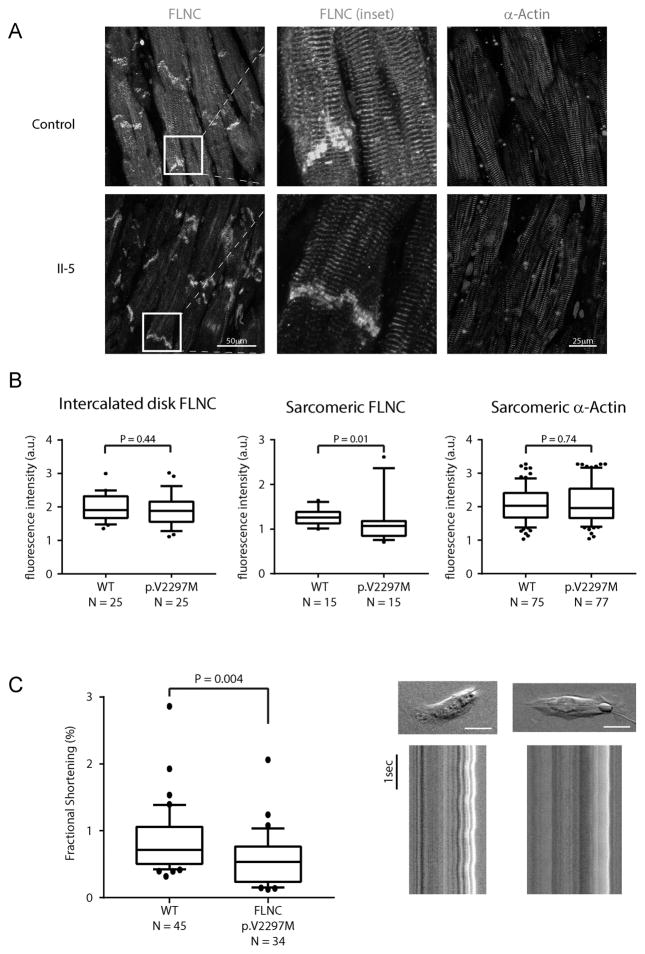
Methods and results: We evaluated 8 family members across 4 generations by history, physical examination, electrocardiography, and echocardiography. Affected individuals presented with a pleitropic syndrome of progressive RCM, atrioventricular septal defects, and a high prevalence of atrial fibrillation. Exome sequencing of 5 affected members identified a single novel missense variant in a highly conserved residue of FLNC (filamin C; p.V2297M). FLNC encodes filamin C-a protein that acts as both a scaffold for the assembly and organization of the central contractile unit of striated muscle and also as a mechanosensitive signaling molecule during cell migration and shear stress. Immunohistochemical analysis of FLNC localization in cardiac tissue from an affected family member revealed a diminished localization at the z disk, whereas traditional localization at the intercalated disk was preserved. Stem cell-derived cardiomyocytes mutated to carry the effect allele had diminished contractile activity when compared with controls.
Conclusion: We have identified a novel variant in FLNC as pathogenic variant for familial RCM-a finding that further expands on the genetic basis of this rare and morbid cardiomyopathy.
Keywords: atrial fibrillation; cardiomyopathy, restrictive; mutation; stem cells; ventricular function.
© 2017 American Heart Association, Inc.
Conflict of interest statement
Dr. Ellinor is a principal investigator on a grant from Bayer Healthcare to study the genetic architecture of atrial fibrillation.
Figures
Comment in
-
FLNC (Filamin-C): A New(er) Player in the Field of Genetic Cardiomyopathies.Circ Cardiovasc Genet. 2017 Dec;10(6):e001959. doi: 10.1161/CIRCGENETICS.117.001959. Circ Cardiovasc Genet. 2017. PMID: 29212901 No abstract available.
-
Letter by Ma et al Regarding Article, "Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy".Circ Genom Precis Med. 2018 Apr;11(4):e002117. doi: 10.1161/CIRCGEN.118.002117. Circ Genom Precis Med. 2018. PMID: 29650767 No abstract available.
-
Response by Ma et al to Letter Regarding Article, "Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy".Circ Genom Precis Med. 2018 Apr;11(4):e002140. doi: 10.1161/CIRCGEN.118.002140. Circ Genom Precis Med. 2018. PMID: 29650770 No abstract available.
References
-
- Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24:214–20. - PubMed
-
- Goldfarb LG, Park KY, Cervenáková L, Gorokhova S, Lee HS, Vasconcelos O, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–3. - PubMed
-
- Kaski JP, Syrris P, Burch M, Tomé-Esteban M-T, Fenton M, Christiansen M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–84. - PubMed
-
- Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, et al. Cardiac beta-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J. 2007;28:2732–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
