

Clinical and neuropathological features of ALS/FTD with TIA1 mutations
- PMID: 29216908
- PMCID: PMC5719900
- DOI: 10.1186/s40478-017-0493-x
Clinical and neuropathological features of ALS/FTD with TIA1 mutations
Abstract
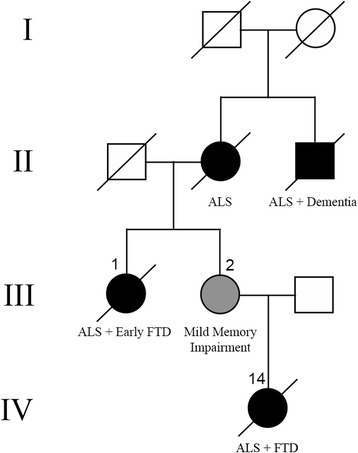
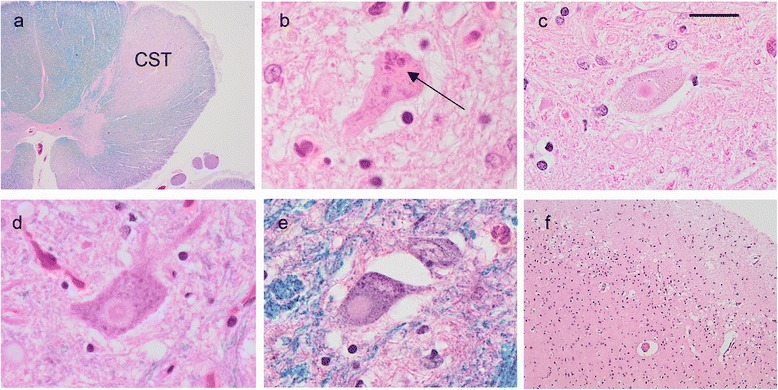
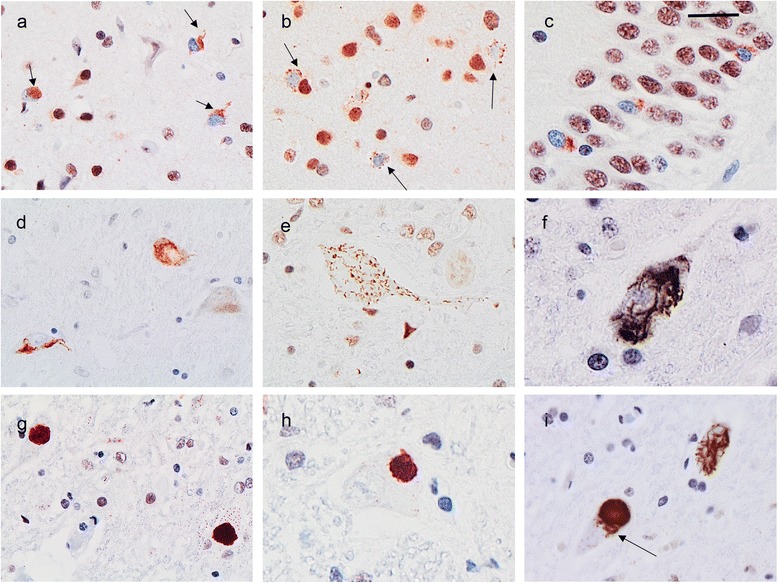
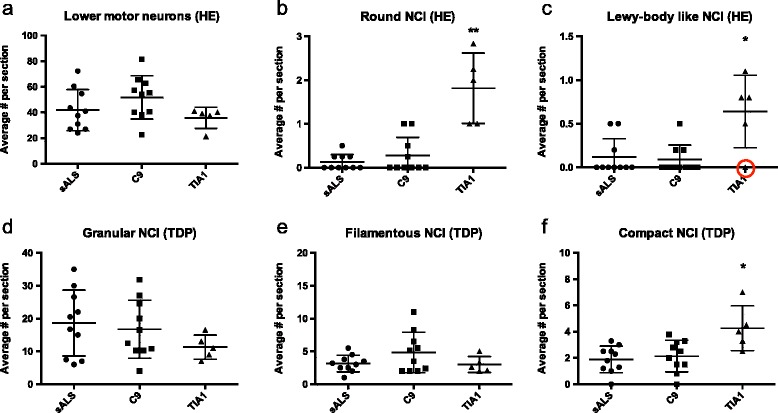
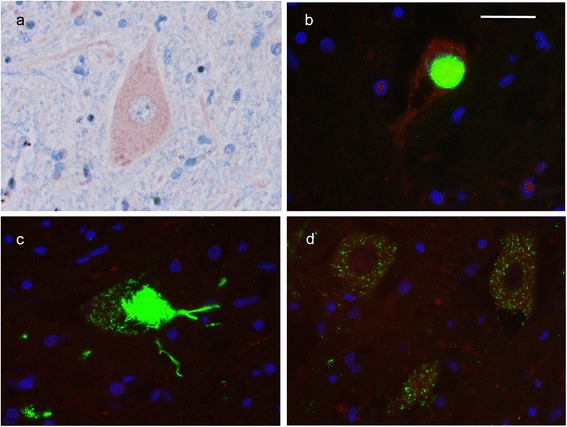
Mutations in the stress granule protein T-cell restricted intracellular antigen 1 (TIA1) were recently shown to cause amyotrophic lateral sclerosis (ALS) with or without frontotemporal dementia (FTD). Here, we provide detailed clinical and neuropathological descriptions of nine cases with TIA1 mutations, together with comparisons to sporadic ALS (sALS) and ALS due to repeat expansions in C9orf72 (C9orf72+). All nine patients with confirmed mutations in TIA1 were female. The clinical phenotype was heterogeneous with a range in the age at onset from late twenties to the eighth decade (mean = 60 years) and disease duration from one to 6 years (mean = 3 years). Initial presentation was either focal weakness or language impairment. All affected individuals received a final diagnosis of ALS with or without FTD. No psychosis or parkinsonism was described. Neuropathological examination on five patients found typical features of ALS and frontotemporal lobar degeneration (FTLD-TDP, type B) with anatomically widespread TDP-43 proteinopathy. In contrast to C9orf72+ cases, caudate atrophy and hippocampal sclerosis were not prominent. Detailed evaluation of the pyramidal motor system found a similar degree of neurodegeneration and TDP-43 pathology as in sALS and C9orf72+ cases; however, cases with TIA1 mutations had increased numbers of lower motor neurons containing round eosinophilic and Lewy body-like inclusions on HE stain and round compact cytoplasmic inclusions with TDP-43 immunohistochemistry. Immunohistochemistry and immunofluorescence failed to demonstrate any labeling of inclusions with antibodies against TIA1. In summary, our TIA1 mutation carriers developed ALS with or without FTD, with a wide range in age at onset, but without other neurological or psychiatric features. The neuropathology was characterized by widespread TDP-43 pathology, but a more restricted pattern of neurodegeneration than C9orf72+ cases. Increased numbers of round eosinophilic and Lewy-body like inclusions in lower motor neurons may be a distinctive feature of ALS caused by TIA1 mutations.
Keywords: Amyotrophic lateral sclerosis; Frontotemporal dementia; Frontotemporal lobar degeneration; T-cell restricted intracellular antigen-1; TDP-43.
Conflict of interest statement
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics.Neuron. 2017 Aug 16;95(4):808-816.e9. doi: 10.1016/j.neuron.2017.07.025. Neuron. 2017. PMID: 28817800 Free PMC article.
-
Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72.Acta Neuropathol. 2011 Dec;122(6):673-90. doi: 10.1007/s00401-011-0907-y. Epub 2011 Nov 15. Acta Neuropathol. 2011. PMID: 22083254 Free PMC article.
-
Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p.Brain. 2012 Mar;135(Pt 3):709-22. doi: 10.1093/brain/awr354. Epub 2012 Feb 17. Brain. 2012. PMID: 22344582 Free PMC article.
-
Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion.Behav Neurol. 2019 Jan 15;2019:2909168. doi: 10.1155/2019/2909168. eCollection 2019. Behav Neurol. 2019. PMID: 30774737 Free PMC article. Review.
-
Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: a systematic neuropathological review.Neuropathol Appl Neurobiol. 2016 Oct;42(6):547-60. doi: 10.1111/nan.12284. Neuropathol Appl Neurobiol. 2016. PMID: 26373655
Cited by
-
Aggregation of the nucleic acid-binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation.J Biol Chem. 2019 Mar 8;294(10):3696-3706. doi: 10.1074/jbc.RA118.006351. Epub 2019 Jan 10. J Biol Chem. 2019. PMID: 30630951 Free PMC article.
-
Hippocampal aggregation signatures of pathogenic UBQLN2 in amyotrophic lateral sclerosis and frontotemporal dementia.Brain. 2024 Oct 3;147(10):3547-3561. doi: 10.1093/brain/awae140. Brain. 2024. PMID: 38703371 Free PMC article.
-
Huntington's disease mice and human brain tissue exhibit increased G3BP1 granules and TDP43 mislocalization.J Clin Invest. 2021 Jun 15;131(12):e140723. doi: 10.1172/JCI140723. J Clin Invest. 2021. PMID: 33945510 Free PMC article.
-
Female sex mitigates motor and behavioural phenotypes in TDP-43Q331K knock-in mice.Sci Rep. 2020 Nov 5;10(1):19220. doi: 10.1038/s41598-020-76070-w. Sci Rep. 2020. PMID: 33154447 Free PMC article.
-
Stress Granules as Causes and Consequences of Translation Suppression.Antioxid Redox Signal. 2023 Aug;39(4-6):390-409. doi: 10.1089/ars.2022.0164. Epub 2023 Jun 28. Antioxid Redox Signal. 2023. PMID: 37183403 Free PMC article. Review.
References
-
- Brooks BR, Miller RG, Swash M, Munsat TL (2000) World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. pp 293–299 - PubMed
-
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, Epstein J, Chiang A, Diaz Z, Nakaya T, Ibrahim F, Kim H-J, Solski JA, Williams KL, Mojsilovic-Petrovic J, Ingre C, Boylan K, Graff-Radford NR, Dickson DW, Clay-Falcone D, Elman L, McCluskey L, Greene R, Kalb RG, Lee VMY, Trojanowski JQ, Ludolph A, Robberecht W, Andersen PM, Nicholson GA, Blair IP, King OD, Bonini NM, Van Deerlin V, Rademakers R, Mourelatos Z, Gitler AD. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
