

Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy
- PMID: 29217642
- PMCID: PMC6954688
- DOI: 10.1161/CIRCULATIONAHA.116.026099
Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy
Erratum in
-
Correction to: Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy.Circulation. 2018 May 22;137(21):e690. doi: 10.1161/CIR.0000000000000581. Circulation. 2018. PMID: 29784693 No abstract available.
Abstract
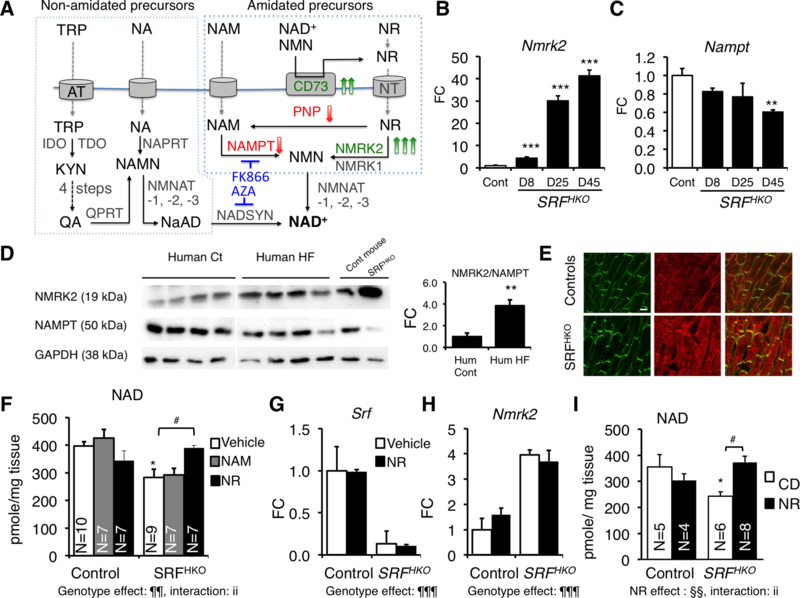
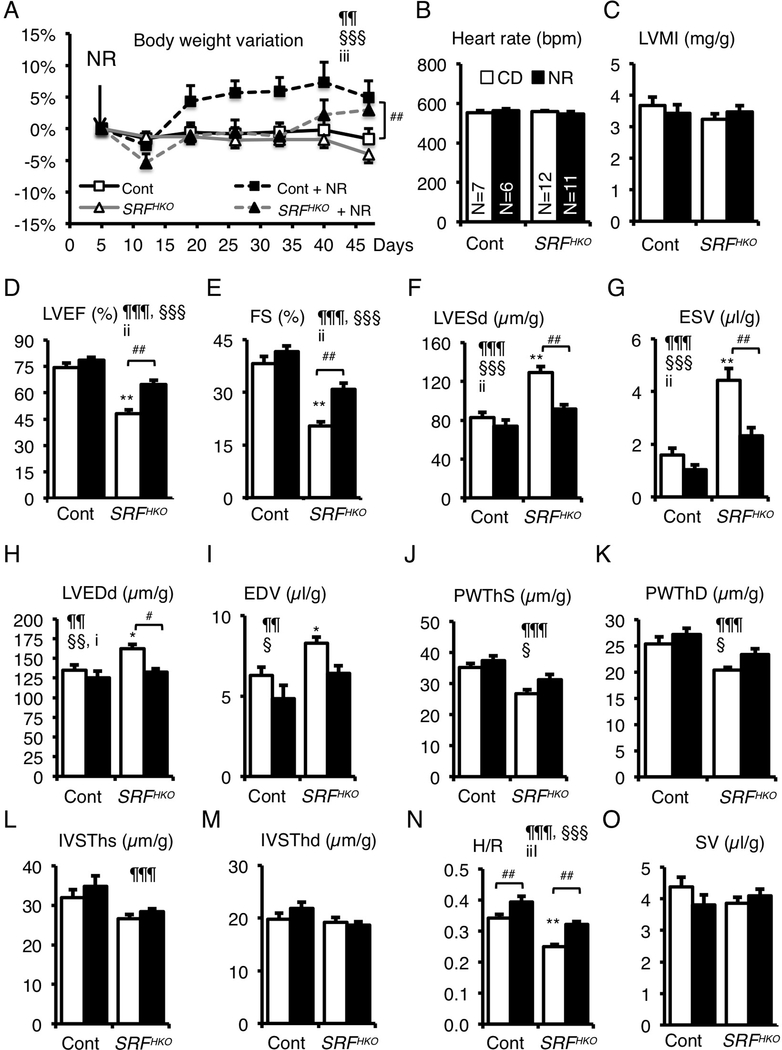
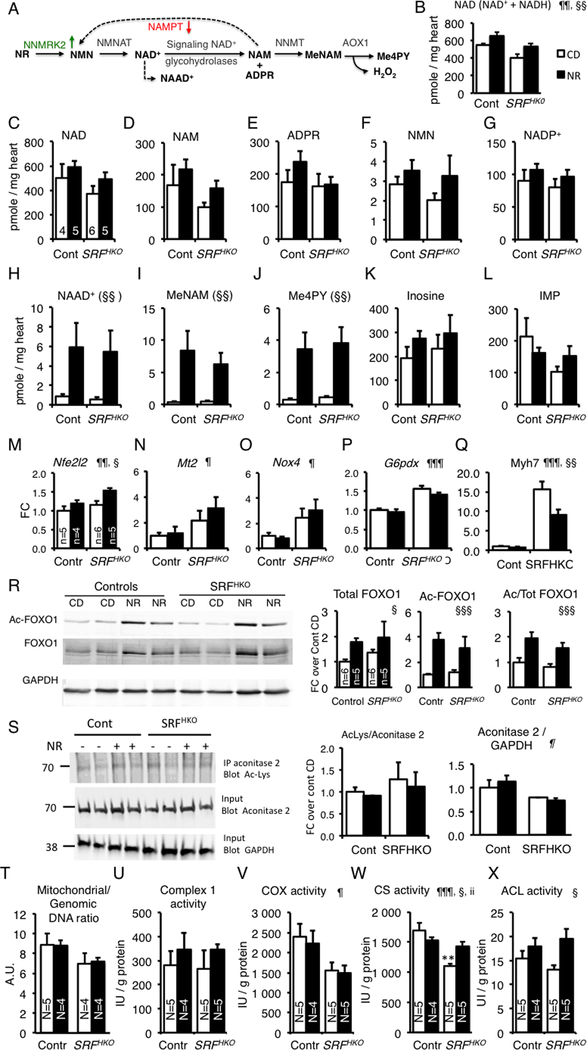
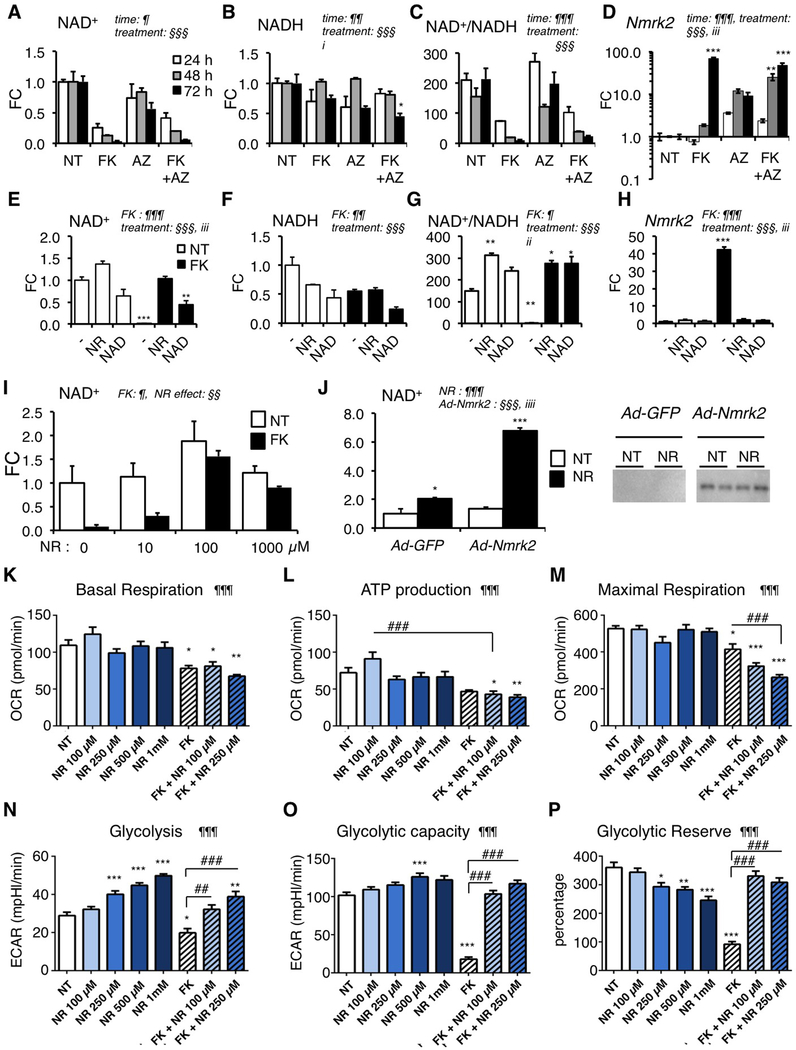
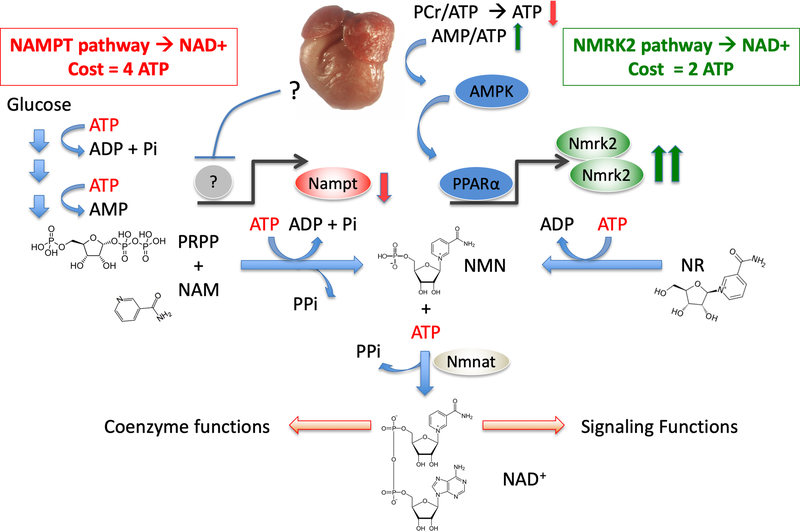
Background: Myocardial metabolic impairment is a major feature in chronic heart failure. As the major coenzyme in fuel oxidation and oxidative phosphorylation and a substrate for enzymes signaling energy stress and oxidative stress response, nicotinamide adenine dinucleotide (NAD+) is emerging as a metabolic target in a number of diseases including heart failure. Little is known on the mechanisms regulating homeostasis of NAD+ in the failing heart.
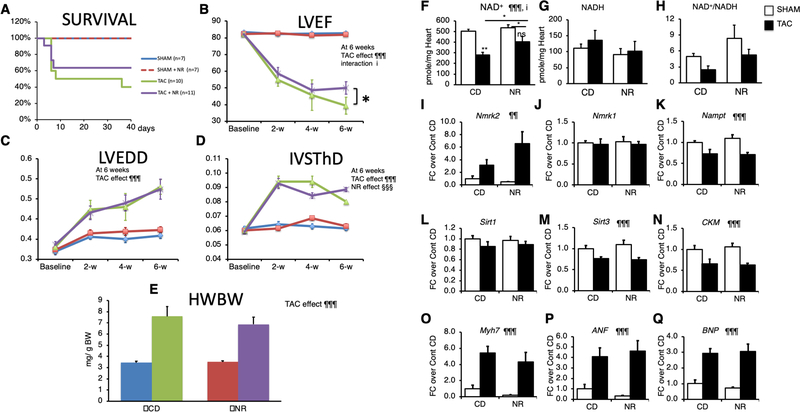
Methods: To explore possible alterations of NAD+ homeostasis in the failing heart, we quantified the expression of NAD+ biosynthetic enzymes in the human failing heart and in the heart of a mouse model of dilated cardiomyopathy (DCM) triggered by Serum Response Factor transcription factor depletion in the heart (SRFHKO) or of cardiac hypertrophy triggered by transverse aorta constriction. We studied the impact of NAD+ precursor supplementation on cardiac function in both mouse models.
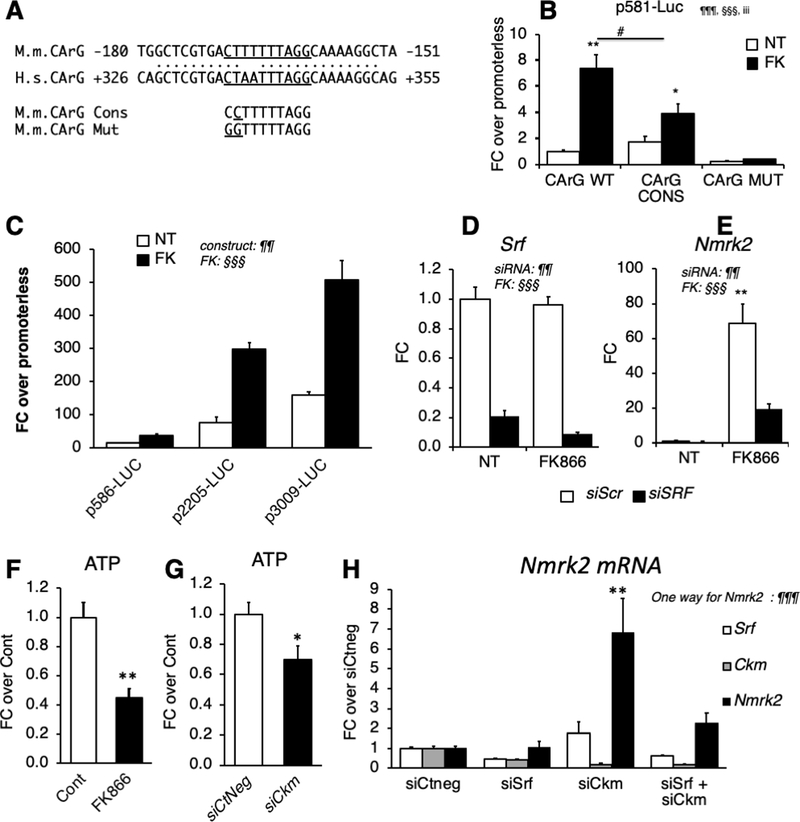
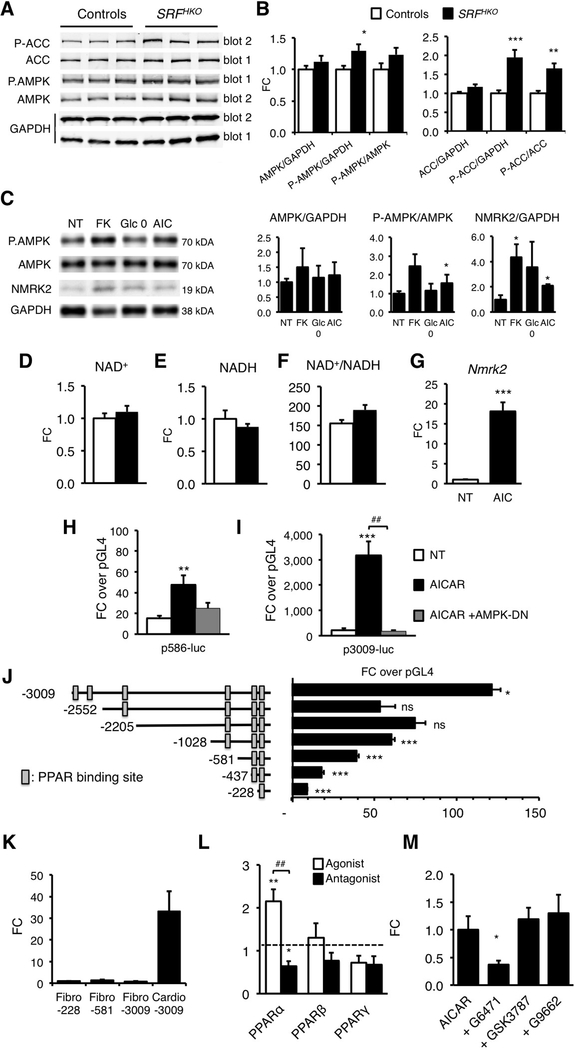
Results: We observed a 30% loss in levels of NAD+ in the murine failing heart of both DCM and transverse aorta constriction mice that was accompanied by a decrease in expression of the nicotinamide phosphoribosyltransferase enzyme that recycles the nicotinamide precursor, whereas the nicotinamide riboside kinase 2 (NMRK2) that phosphorylates the nicotinamide riboside precursor is increased, to a higher level in the DCM (40-fold) than in transverse aorta constriction (4-fold). This shift was also observed in human failing heart biopsies in comparison with nonfailing controls. We show that the Nmrk2 gene is an AMP-activated protein kinase and peroxisome proliferator-activated receptor α responsive gene that is activated by energy stress and NAD+ depletion in isolated rat cardiomyocytes. Nicotinamide riboside efficiently rescues NAD+ synthesis in response to FK866-mediated inhibition of nicotinamide phosphoribosyltransferase and stimulates glycolysis in cardiomyocytes. Accordingly, we show that nicotinamide riboside supplementation in food attenuates the development of heart failure in mice, more robustly in DCM, and partially after transverse aorta constriction, by stabilizing myocardial NAD+ levels in the failing heart. Nicotinamide riboside treatment also robustly increases the myocardial levels of 3 metabolites, nicotinic acid adenine dinucleotide, methylnicotinamide, and N1-methyl-4-pyridone-5-carboxamide, that can be used as validation biomarkers for the treatment.
Conclusions: The data show that nicotinamide riboside, the most energy-efficient among NAD precursors, could be useful for treatment of heart failure, notably in the context of DCM, a disease with few therapeutic options.
Keywords: NAD; acetyl coenzyme A; cardiomyopathy, dilated; energy metabolism; glycolysis; heart failure; nicotinamide-beta-riboside; serum response factor.
© 2017 American Heart Association, Inc.
Figures
Comment in
-
Raising NAD in Heart Failure: Time to Translate?Circulation. 2018 May 22;137(21):2274-2277. doi: 10.1161/CIRCULATIONAHA.117.032626. Circulation. 2018. PMID: 29784680 Free PMC article. No abstract available.
References
-
- Ventura-Clapier R, Garnier A, Veksler V and Joubert F. Bioenergetics of the failing heart. Biochim Biophys Acta. 2011;1813:1360–72. - PubMed
-
- Mericskay M Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis. 2016;109:207–15. - PubMed
-
- Lee CF and Tian R. Mitochondrion as a Target for Heart Failure Therapy- Role of Protein Lysine Acetylation. Circulation journal : official journal of the Japanese Circulation Society. 2015;79:1863–70. - PubMed
-
- Belenky P, Bogan KL and Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
