

Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7
- PMID: 29217778
- PMCID: PMC5830370
- DOI: 10.3324/haematol.2017.180778
Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7
Abstract
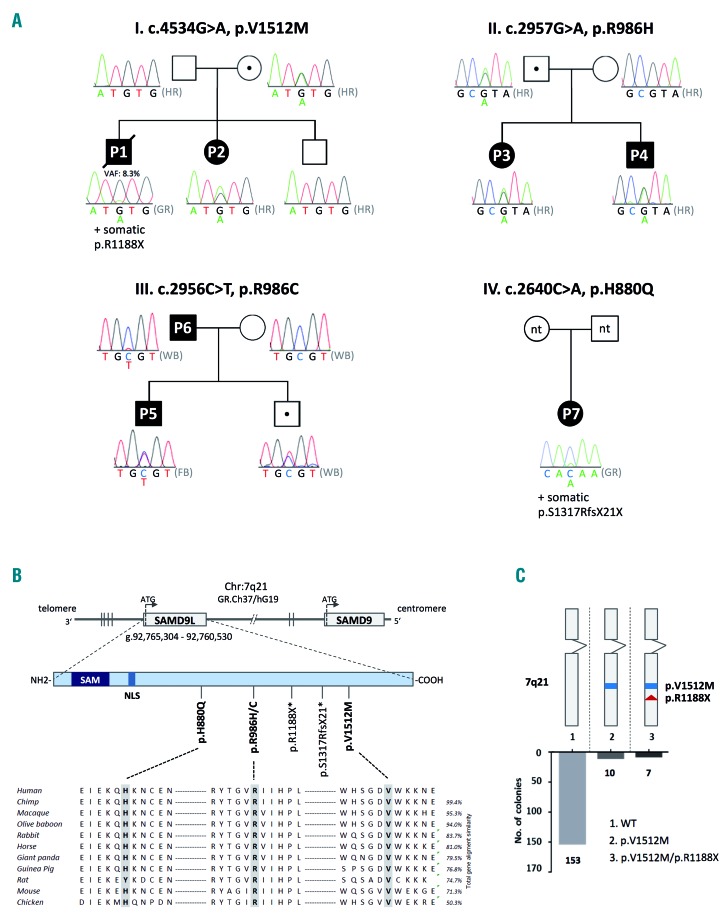
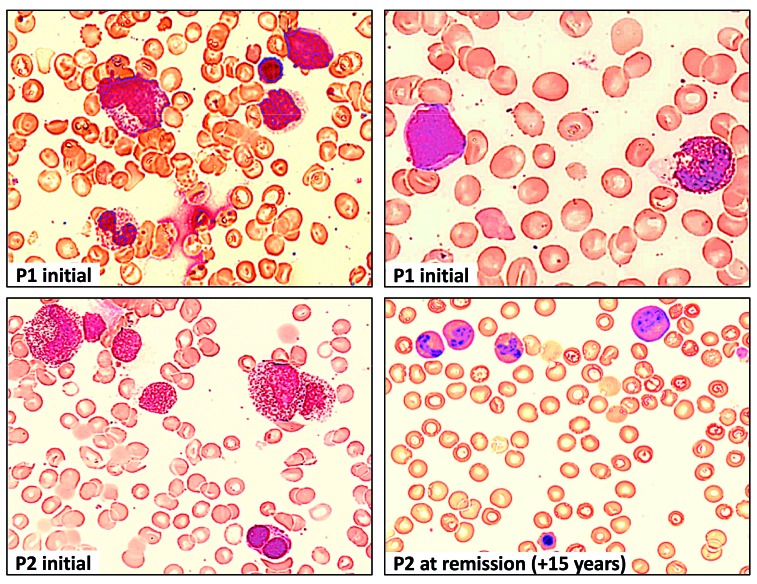
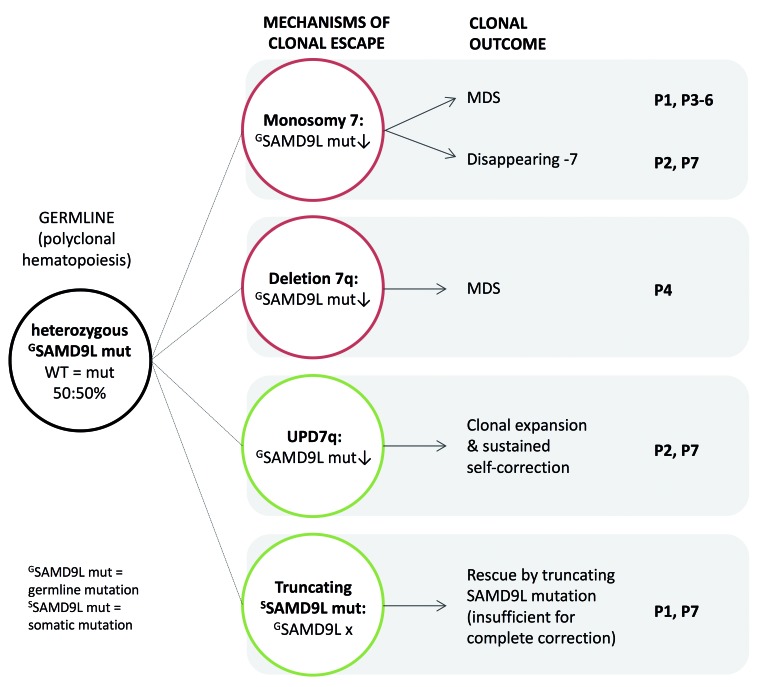
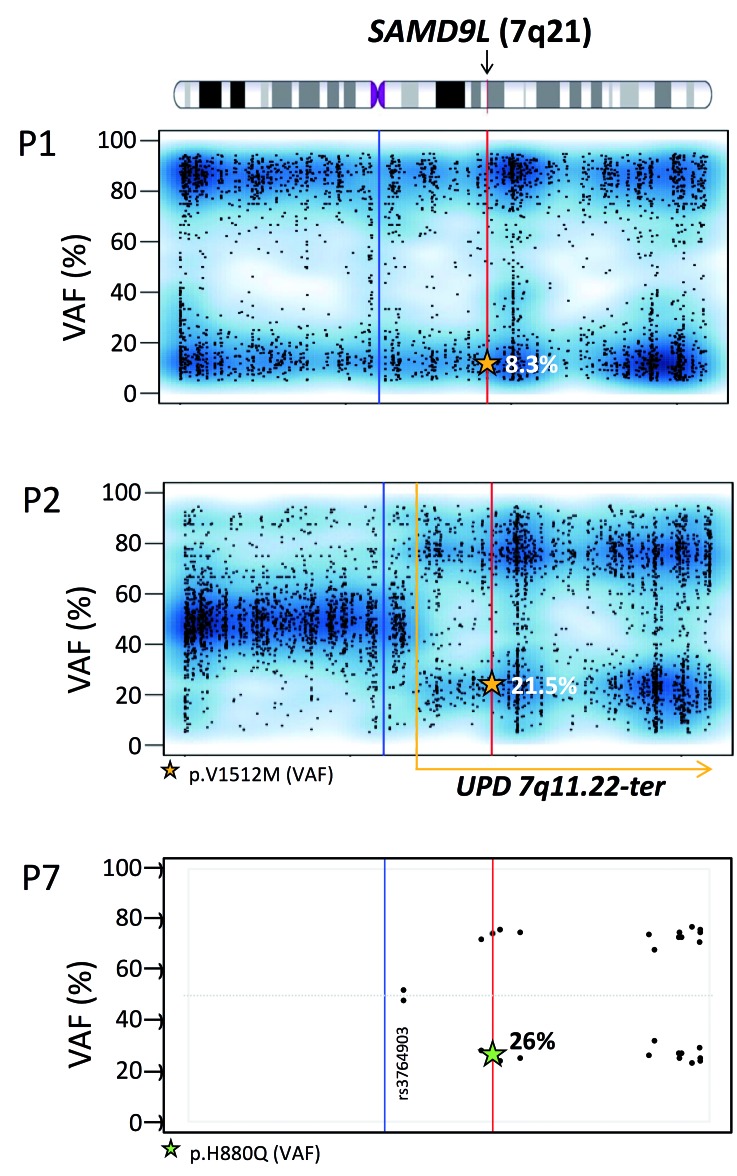
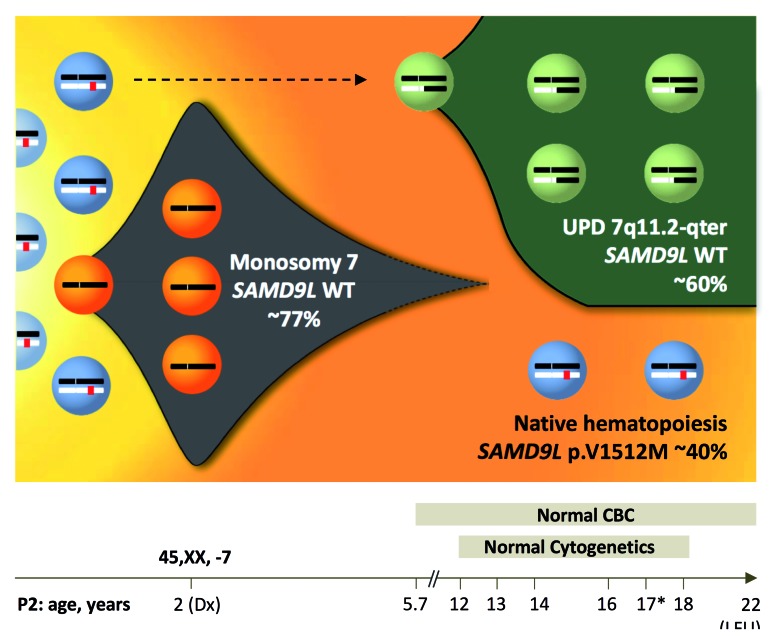
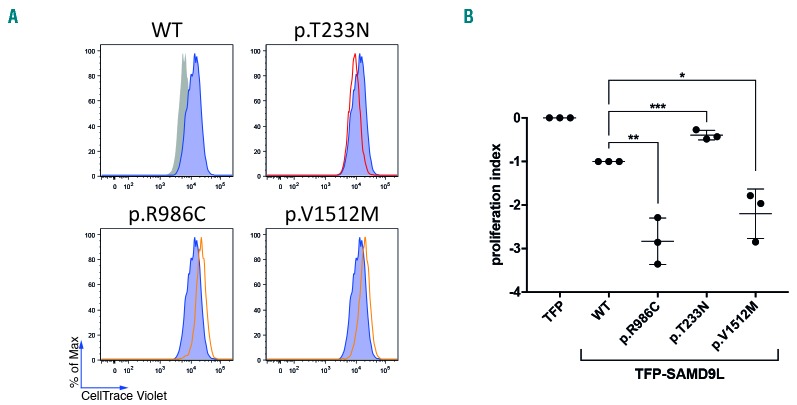
Familial myelodysplastic syndromes arise from haploinsufficiency of genes involved in hematopoiesis and are primarily associated with early-onset disease. Here we describe a familial syndrome in seven patients from four unrelated pedigrees presenting with myelodysplastic syndrome and loss of chromosome 7/7q. Their median age at diagnosis was 2.1 years (range, 1-42). All patients presented with thrombocytopenia with or without additional cytopenias and a hypocellular marrow without an increase of blasts. Genomic studies identified constitutional mutations (p.H880Q, p.R986H, p.R986C and p.V1512M) in the SAMD9L gene on 7q21, with decreased allele frequency in hematopoiesis. The non-random loss of mutated SAMD9L alleles was attained via monosomy 7, deletion 7q, UPD7q, or acquired truncating SAMD9L variants p.R1188X and p.S1317RfsX21. Incomplete penetrance was noted in 30% (3/10) of mutation carriers. Long-term observation revealed divergent outcomes with either progression to leukemia and/or accumulation of driver mutations (n=2), persistent monosomy 7 (n=4), and transient monosomy 7 followed by spontaneous recovery with SAMD9L-wildtype UPD7q (n=2). Dysmorphic features or neurological symptoms were absent in our patients, pointing to the notion that myelodysplasia with monosomy 7 can be a sole manifestation of SAMD9L disease. Collectively, our results define a new subtype of familial myelodysplastic syndrome and provide an explanation for the phenomenon of transient monosomy 7. Registered at: www.clinicaltrials.gov; #NCT00047268.
Copyright© 2018 Ferrata Storti Foundation.
Figures
References
-
- Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43(5):598–608. - PubMed
-
- Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403–2407. - PubMed
-
- Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
