

Large-scale analysis of disease pathways in the human interactome
- PMID: 29218874
- PMCID: PMC5731453
Large-scale analysis of disease pathways in the human interactome
Abstract
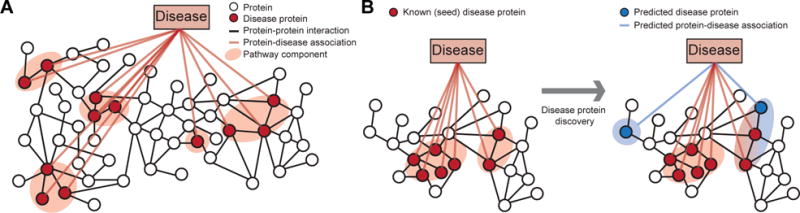
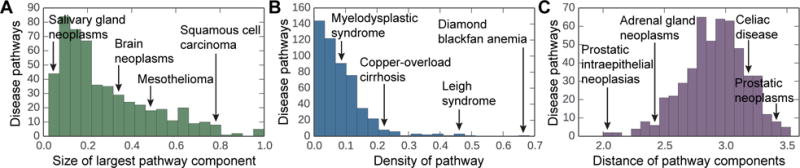
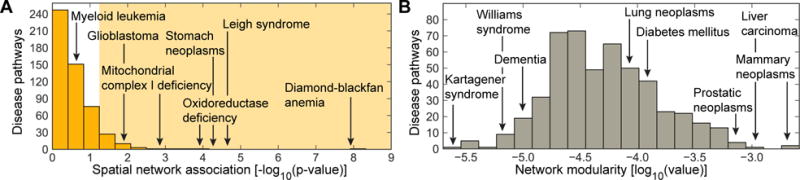
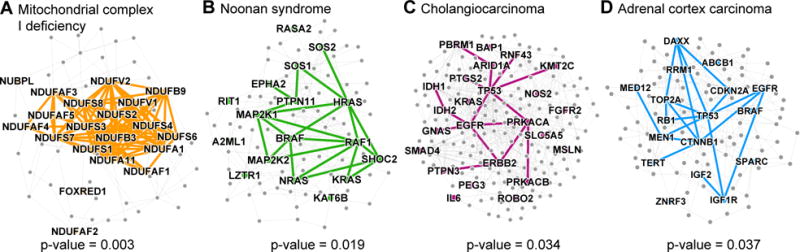
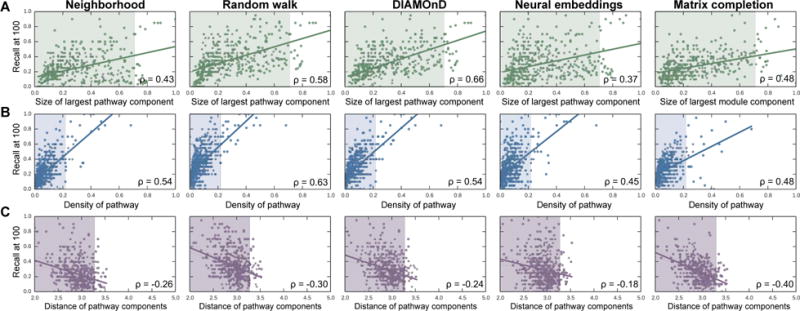
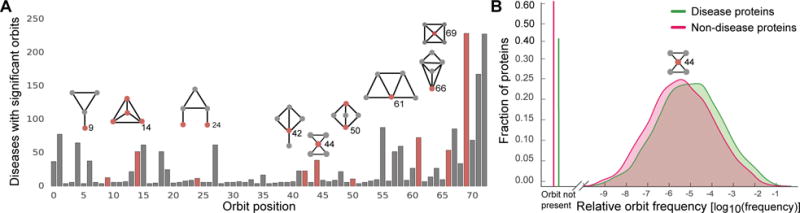
Discovering disease pathways, which can be defined as sets of proteins associated with a given disease, is an important problem that has the potential to provide clinically actionable insights for disease diagnosis, prognosis, and treatment. Computational methods aid the discovery by relying on protein-protein interaction (PPI) networks. They start with a few known disease-associated proteins and aim to find the rest of the pathway by exploring the PPI network around the known disease proteins. However, the success of such methods has been limited, and failure cases have not been well understood. Here we study the PPI network structure of 519 disease pathways. We find that 90% of pathways do not correspond to single well-connected components in the PPI network. Instead, proteins associated with a single disease tend to form many separate connected components/regions in the network. We then evaluate state-of-the-art disease pathway discovery methods and show that their performance is especially poor on diseases with disconnected pathways. Thus, we conclude that network connectivity structure alone may not be sufficient for disease pathway discovery. However, we show that higher-order network structures, such as small subgraphs of the pathway, provide a promising direction for the development of new methods.
Figures
References
-
- Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Nature Reviews Genetics. 2015;16:85. - PubMed
-
- Piñero J, et al. Database. 2015;2015
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
