

High-Resolution Single-Cell Sequencing of Malaria Parasites
- PMID: 29220419
- PMCID: PMC5737330
- DOI: 10.1093/gbe/evx256
High-Resolution Single-Cell Sequencing of Malaria Parasites
Abstract
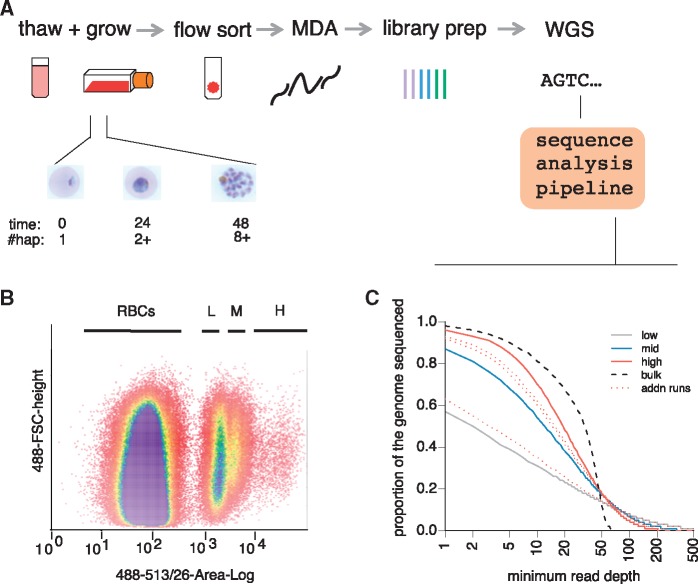
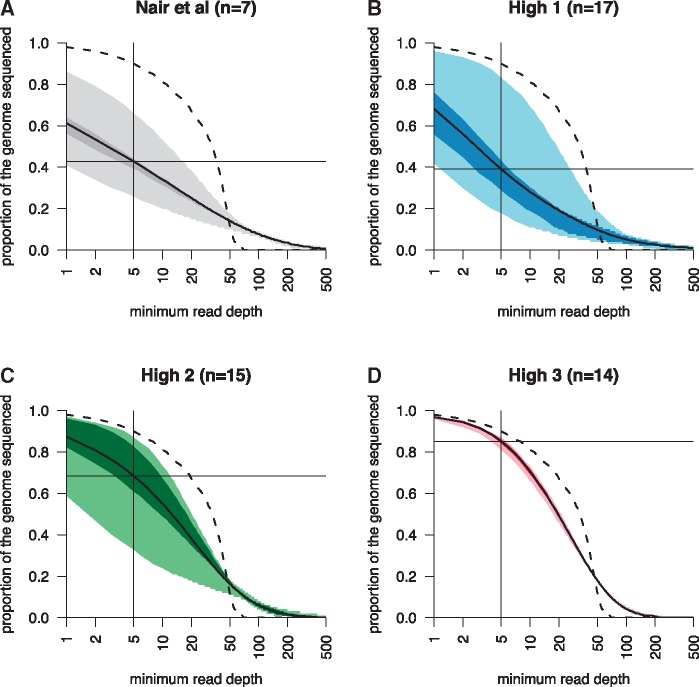
Single-cell genomics is a powerful tool for determining the genetic architecture of complex communities of unicellular organisms. In areas of high transmission, malaria patients are often challenged by the activities of multiple Plasmodium falciparum lineages, which can potentiate pathology, spread drug resistance loci, and also complicate most genetic analysis. Single-cell sequencing of P. falciparum would be key to understanding infection complexity, though efforts are hampered by the extreme nucleotide composition of its genome (∼80% AT-rich). To counter the low coverage achieved in previous studies, we targeted DNA-rich late-stage parasites by Fluorescence-Activated Cell Sorting and whole genome sequencing. Our method routinely generates accurate, near-complete capture of the 23 Mb P. falciparum genome (mean breadth of coverage 90.7%) at high efficiency. Data from 48 single-cell genomes derived from a polyclonal infection sampled in Chikhwawa, Malawi allowed for unambiguous determination of haplotype diversity and recent meiotic events, information that will aid public health efforts.
Keywords: malaria; methods; single-cell genomics.
© The Author(s) 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures




References
-
- Assefa SA, et al.2014. estMOI: estimating multiplicity of infection using parasite deep sequencing data. Bioinformatics 30(9):1292–1294.http://dx.doi.org/10.1093/bioinformatics/btu005 - DOI - PMC - PubMed
-
- Auton A, McVean G.. 2007. Recombination rate estimation in the presence of hotspots. Genome Res. 17(8):1219–1227.http://dx.doi.org/10.1101/gr.6386707 - DOI - PMC - PubMed
-
- Bourgon R. 2015. Intervals: Tools for Working with Points and Intervals.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
