

A Powerful Approach to Estimating Annotation-Stratified Genetic Covariance via GWAS Summary Statistics
- PMID: 29220677
- PMCID: PMC5812911
- DOI: 10.1016/j.ajhg.2017.11.001
A Powerful Approach to Estimating Annotation-Stratified Genetic Covariance via GWAS Summary Statistics
Abstract
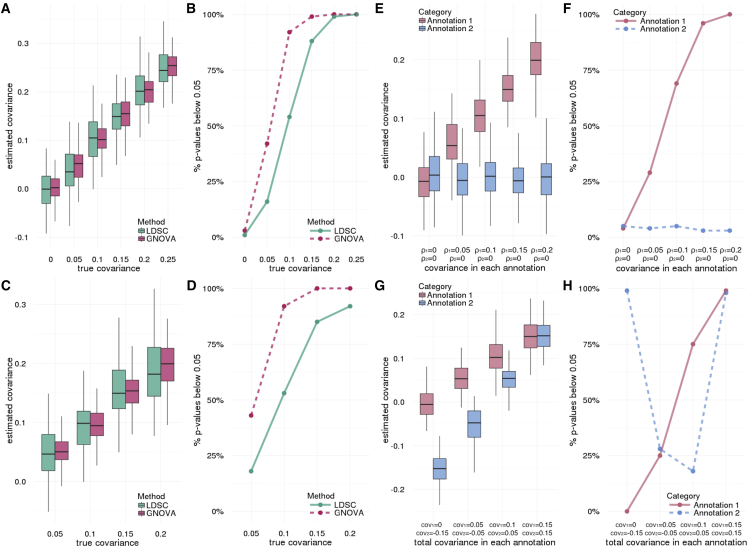
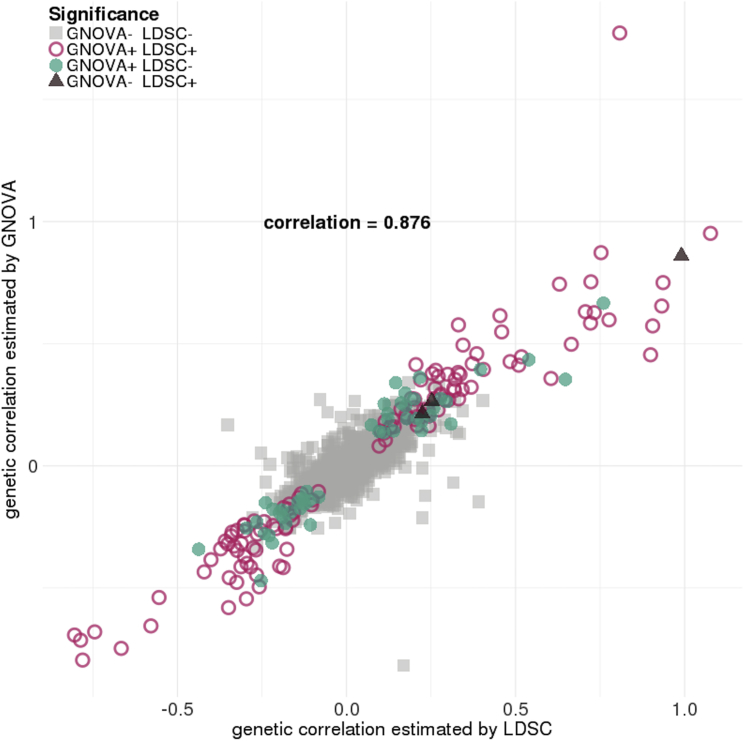
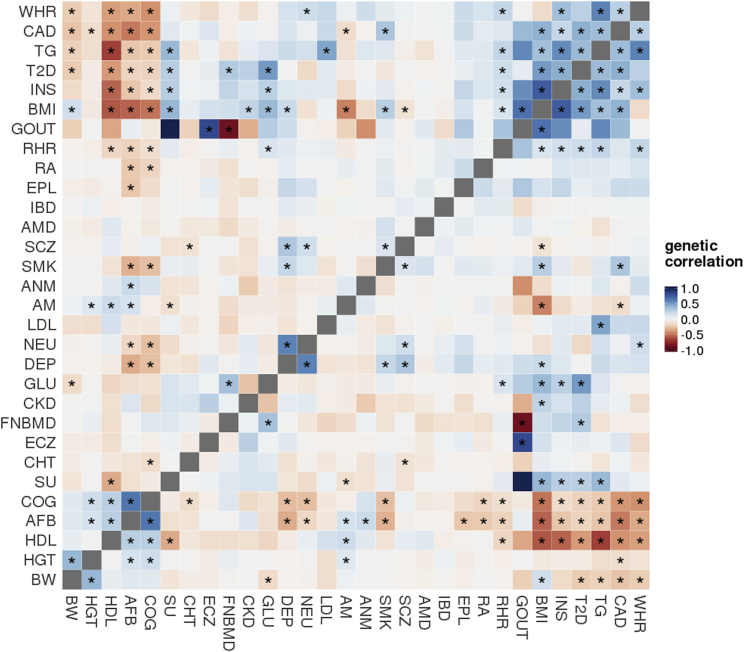
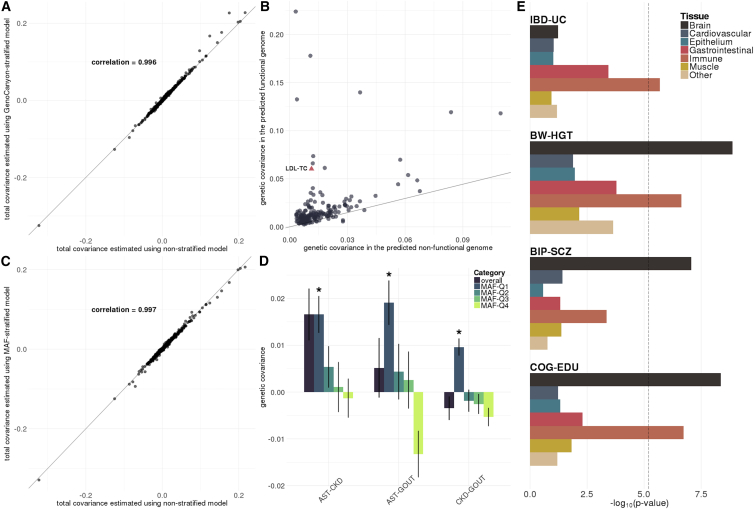
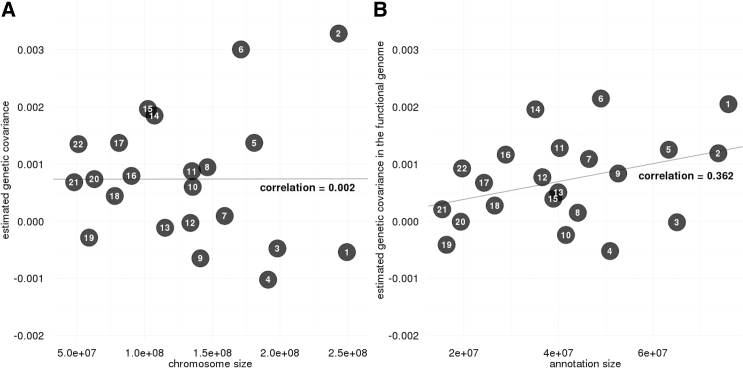
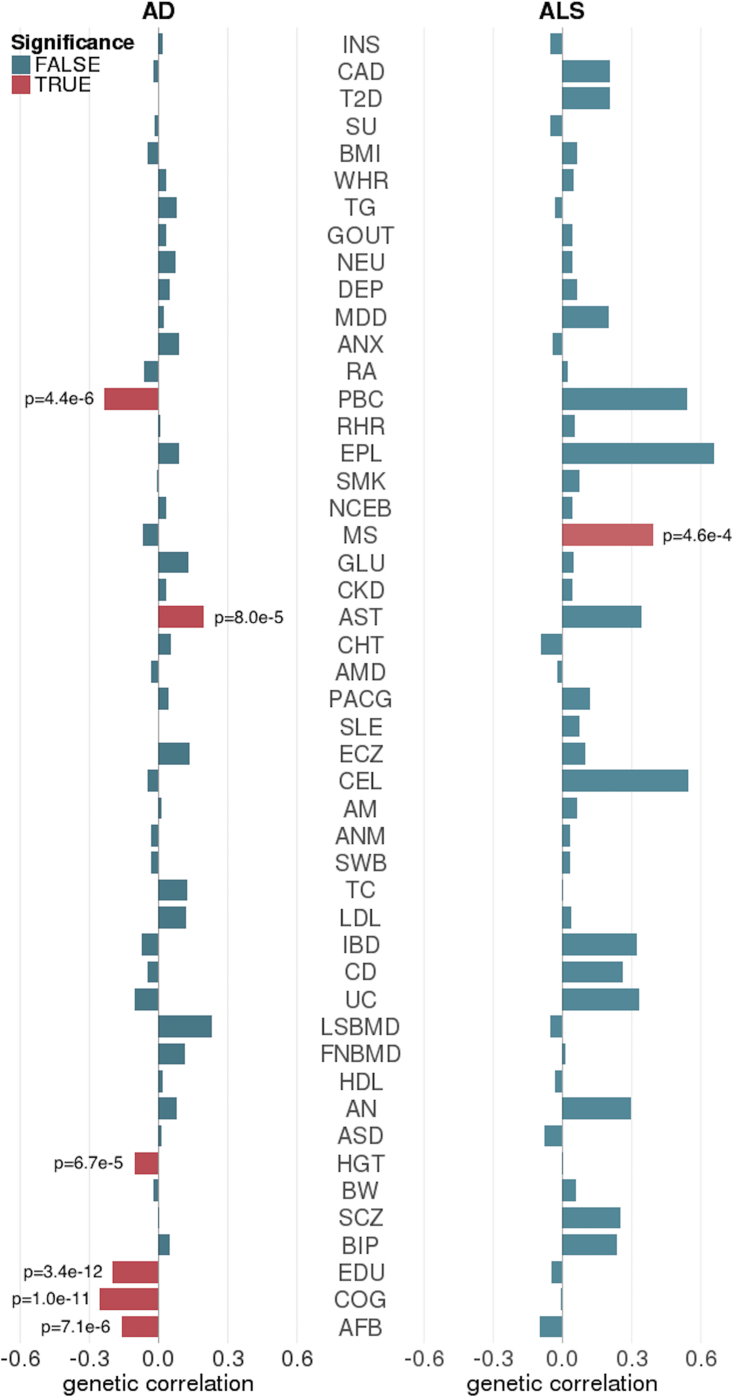
Despite the success of large-scale genome-wide association studies (GWASs) on complex traits, our understanding of their genetic architecture is far from complete. Jointly modeling multiple traits' genetic profiles has provided insights into the shared genetic basis of many complex traits. However, large-scale inference sets a high bar for both statistical power and biological interpretability. Here we introduce a principled framework to estimate annotation-stratified genetic covariance between traits using GWAS summary statistics. Through theoretical and numerical analyses, we demonstrate that our method provides accurate covariance estimates, thereby enabling researchers to dissect both the shared and distinct genetic architecture across traits to better understand their etiologies. Among 50 complex traits with publicly accessible GWAS summary statistics (Ntotal≈ 4.5 million), we identified more than 170 pairs with statistically significant genetic covariance. In particular, we found strong genetic covariance between late-onset Alzheimer disease (LOAD) and amyotrophic lateral sclerosis (ALS), two major neurodegenerative diseases, in single-nucleotide polymorphisms (SNPs) with high minor allele frequencies and in SNPs located in the predicted functional genome. Joint analysis of LOAD, ALS, and other traits highlights LOAD's correlation with cognitive traits and hints at an autoimmune component for ALS.
Keywords: Alzheimer’s disease; amyotrophic lateral sclerosis; functional annotation; genetic covariance; genome-wide association study; summary statistics.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
