

Exploring digenic inheritance in arrhythmogenic cardiomyopathy
- PMID: 29221435
- PMCID: PMC5723071
- DOI: 10.1186/s12881-017-0503-7
Exploring digenic inheritance in arrhythmogenic cardiomyopathy
Abstract
Background: Arrhythmogenic cardiomyopathy (ACM) is an inherited genetic disorder, characterized by the substitution of heart muscle with fibro-fatty tissue and severe ventricular arrhythmias, often leading to heart failure and sudden cardiac death. ACM is considered a monogenic disorder, but the low penetrance of mutations identified in patients suggests the involvement of additional genetic or environmental factors.
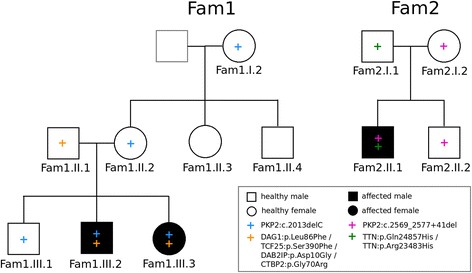
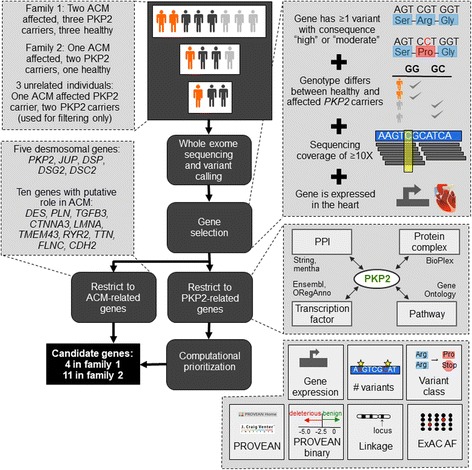
Methods: We used whole exome sequencing to investigate digenic inheritance in two ACM families where previous diagnostic tests have revealed a PKP2 mutation in all affected and some healthy individuals. In family members with PKP2 mutations we determined all genes that harbor variants in affected but not in healthy carriers or vice versa. We computationally prioritized the most likely candidates, focusing on known ACM genes and genes related to PKP2 through protein interactions, functional relationships, or shared biological processes.
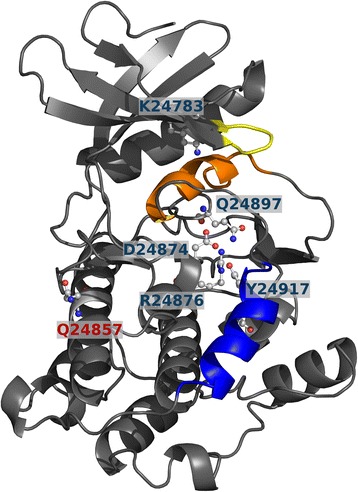
Results: We identified four candidate genes in family 1, namely DAG1, DAB2IP, CTBP2 and TCF25, and eleven candidate genes in family 2. The most promising gene in the second family is TTN, a gene previously associated with ACM, in which the affected individual harbors two rare deleterious-predicted missense variants, one of which is located in the protein's only serine kinase domain.
Conclusions: In this study we report genes that might act as digenic players in ACM pathogenesis, on the basis of co-segregation with PKP2 mutations. Validation in larger cohorts is still required to prove the utility of this model.
Keywords: ACM; Arrhythmogenic cardiomyopathy; Digenic inheritance; Exome sequencing; PKP2.
Conflict of interest statement
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, et al. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. - DOI - PMC - PubMed
-
- Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, et al. Identification of cadherin 2 (CDH2) mutations in Arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10(2) - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
