

Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data
- PMID: 29226803
- PMCID: PMC5799033
- DOI: 10.1016/j.cels.2017.11.003
Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data
Abstract
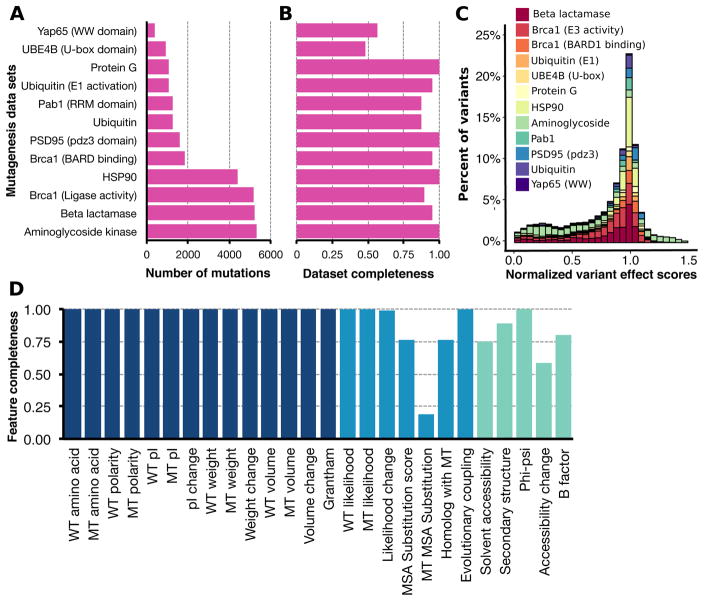
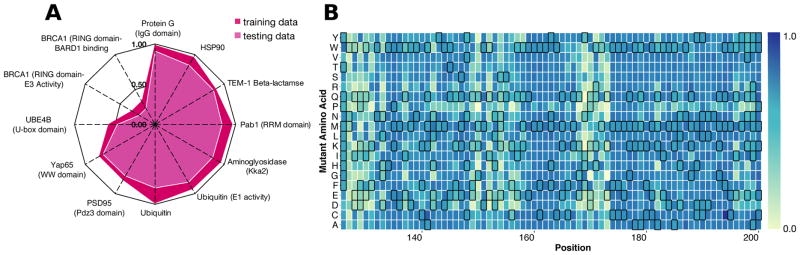
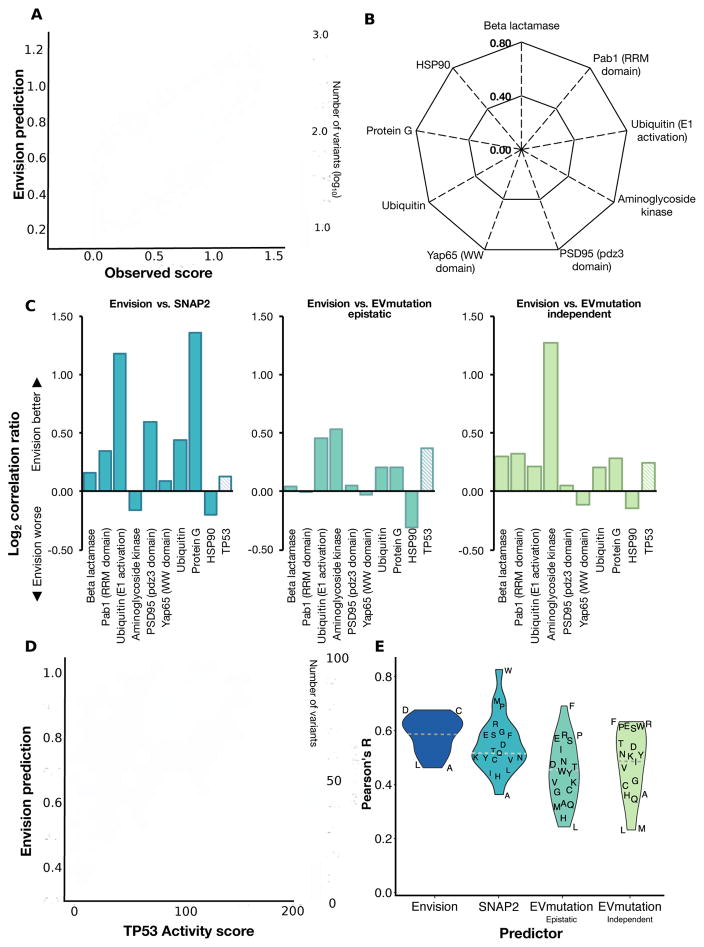
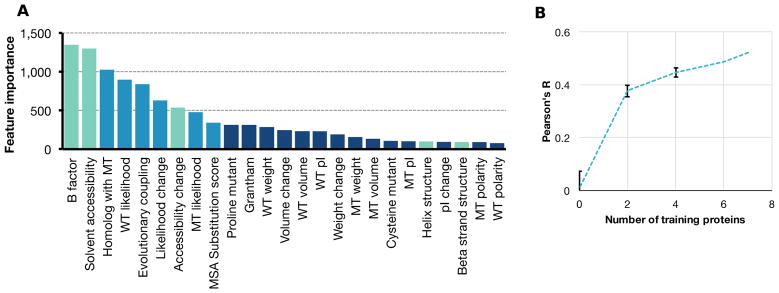
Large datasets describing the quantitative effects of mutations on protein function are becoming increasingly available. Here, we leverage these datasets to develop Envision, which predicts the magnitude of a missense variant's molecular effect. Envision combines 21,026 variant effect measurements from nine large-scale experimental mutagenesis datasets, a hitherto untapped training resource, with a supervised, stochastic gradient boosting learning algorithm. Envision outperforms other missense variant effect predictors both on large-scale mutagenesis data and on an independent test dataset comprising 2,312 TP53 variants whose effects were measured using a low-throughput approach. This dataset was never used for hyperparameter tuning or model training and thus serves as an independent validation set. Envision prediction accuracy is also more consistent across amino acids than other predictors. Finally, we demonstrate that Envision's performance improves as more large-scale mutagenesis data are incorporated. We precompute Envision predictions for every possible single amino acid variant in human, mouse, frog, zebrafish, fruit fly, worm, and yeast proteomes (https://envision.gs.washington.edu/).
Keywords: large-scale mutagenesis; machine learning; variant effect prediction.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System, the 22nd ACM SIGKDD International Conference; New York, New York, USA: ACM; 2016. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
