

Drug-perturbation-based stratification of blood cancer
- PMID: 29227286
- PMCID: PMC5749541
- DOI: 10.1172/JCI93801
Drug-perturbation-based stratification of blood cancer
Abstract
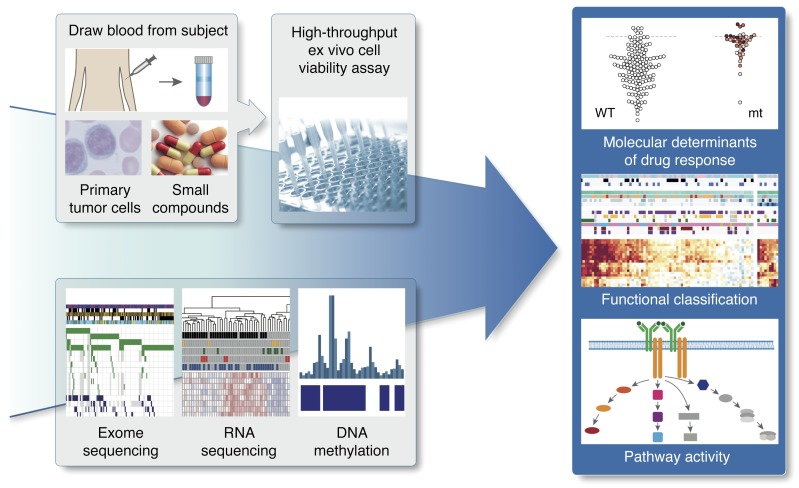
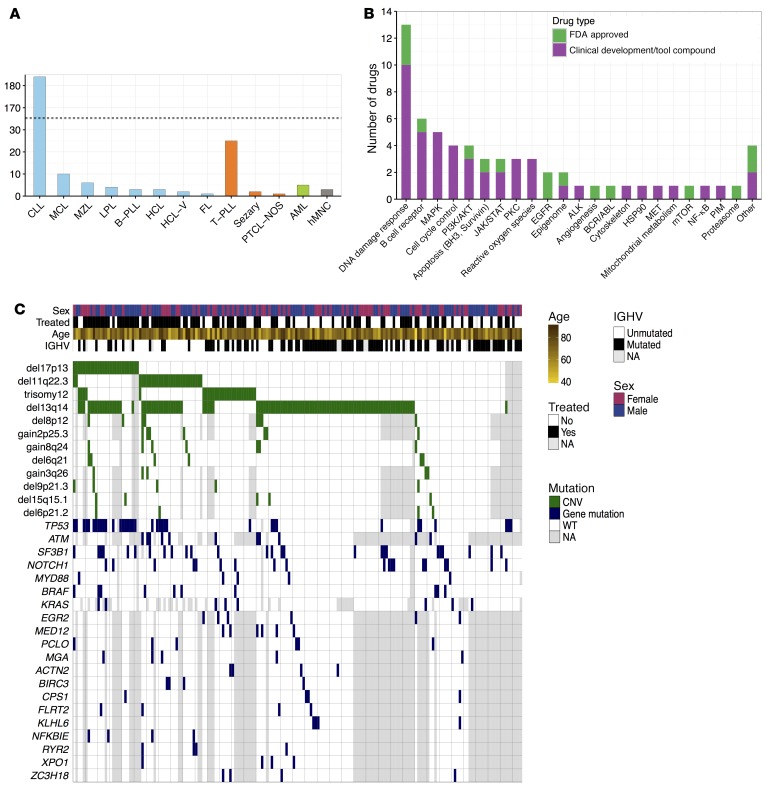
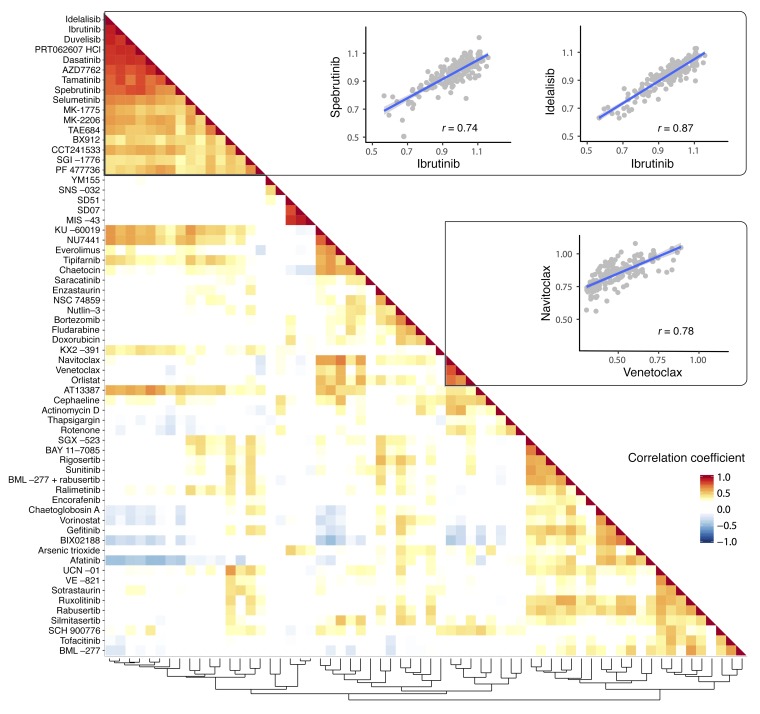
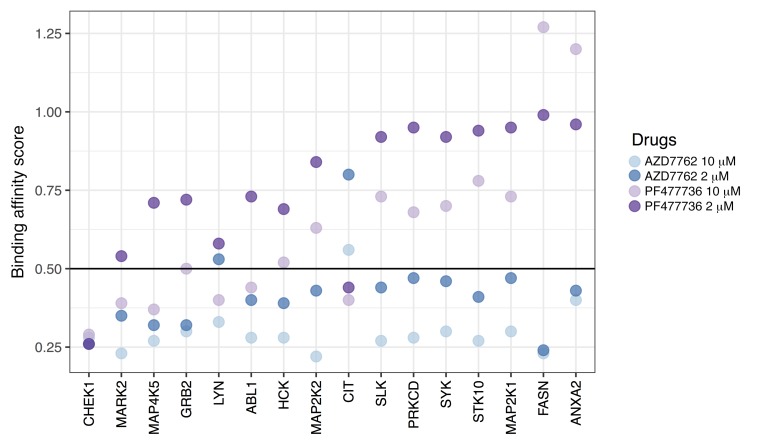
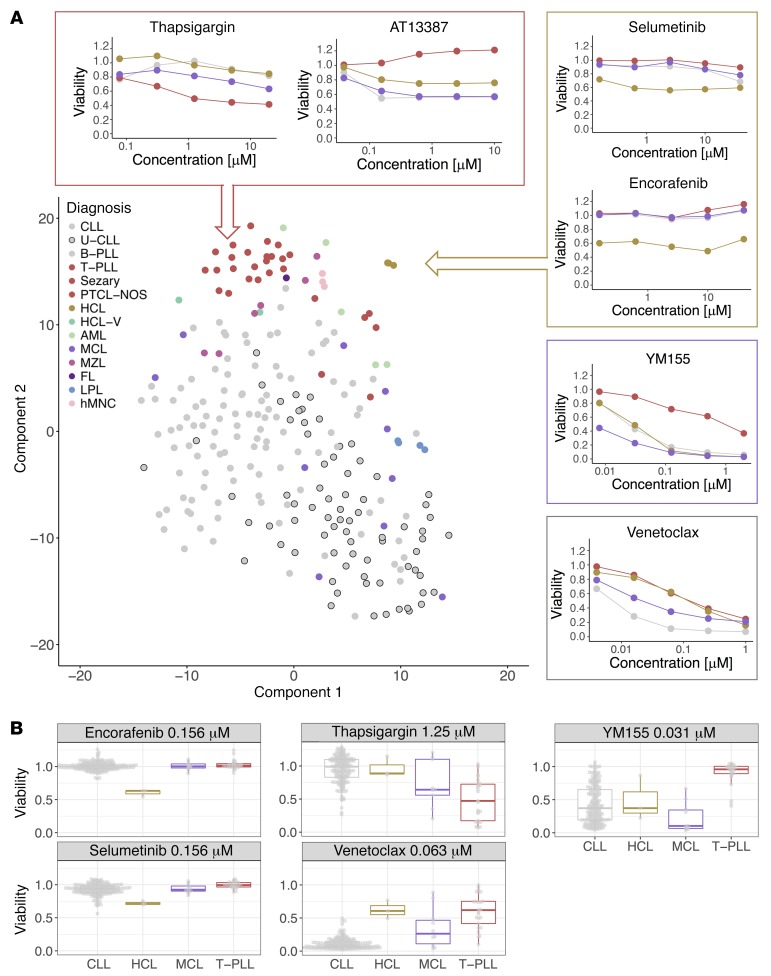
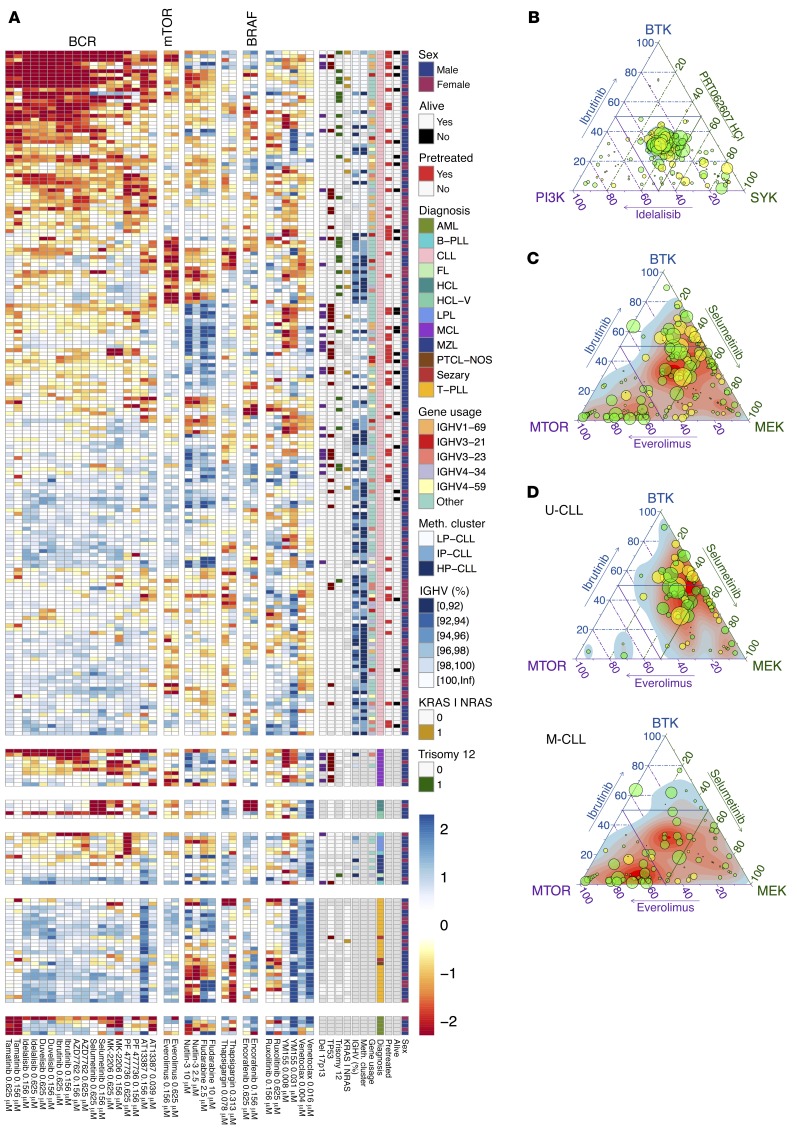
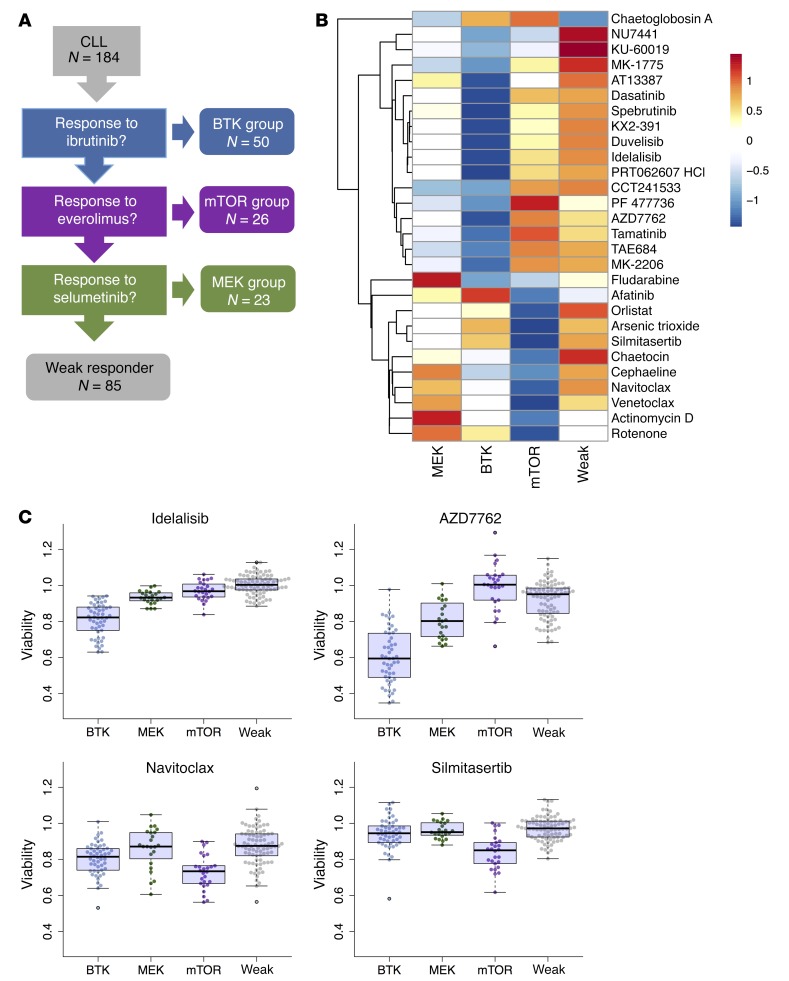
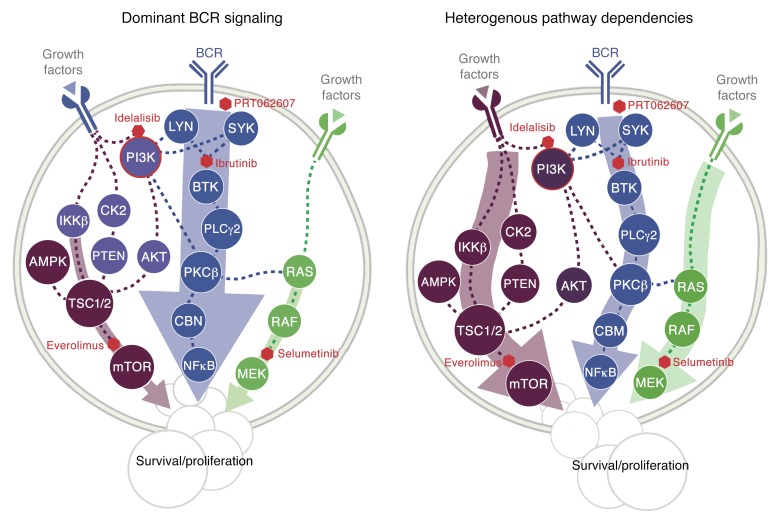
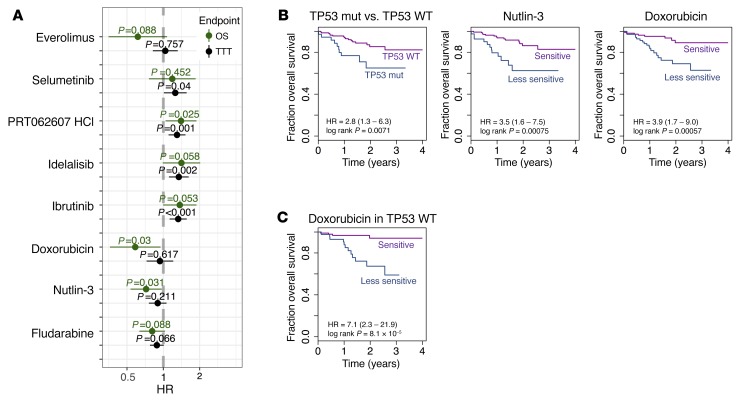
As new generations of targeted therapies emerge and tumor genome sequencing discovers increasingly comprehensive mutation repertoires, the functional relationships of mutations to tumor phenotypes remain largely unknown. Here, we measured ex vivo sensitivity of 246 blood cancers to 63 drugs alongside genome, transcriptome, and DNA methylome analysis to understand determinants of drug response. We assembled a primary blood cancer cell encyclopedia data set that revealed disease-specific sensitivities for each cancer. Within chronic lymphocytic leukemia (CLL), responses to 62% of drugs were associated with 2 or more mutations, and linked the B cell receptor (BCR) pathway to trisomy 12, an important driver of CLL. Based on drug responses, the disease could be organized into phenotypic subgroups characterized by exploitable dependencies on BCR, mTOR, or MEK signaling and associated with mutations, gene expression, and DNA methylation. Fourteen percent of CLLs were driven by mTOR signaling in a non-BCR-dependent manner. Multivariate modeling revealed immunoglobulin heavy chain variable gene (IGHV) mutation status and trisomy 12 as the most important modulators of response to kinase inhibitors in CLL. Ex vivo drug responses were associated with outcome. This study overcomes the perception that most mutations do not influence drug response of cancer, and points to an updated approach to understanding tumor biology, with implications for biomarker discovery and cancer care.
Keywords: B cell receptor; Drug screens; Hematology; Leukemias; Oncology.
Conflict of interest statement
Figures
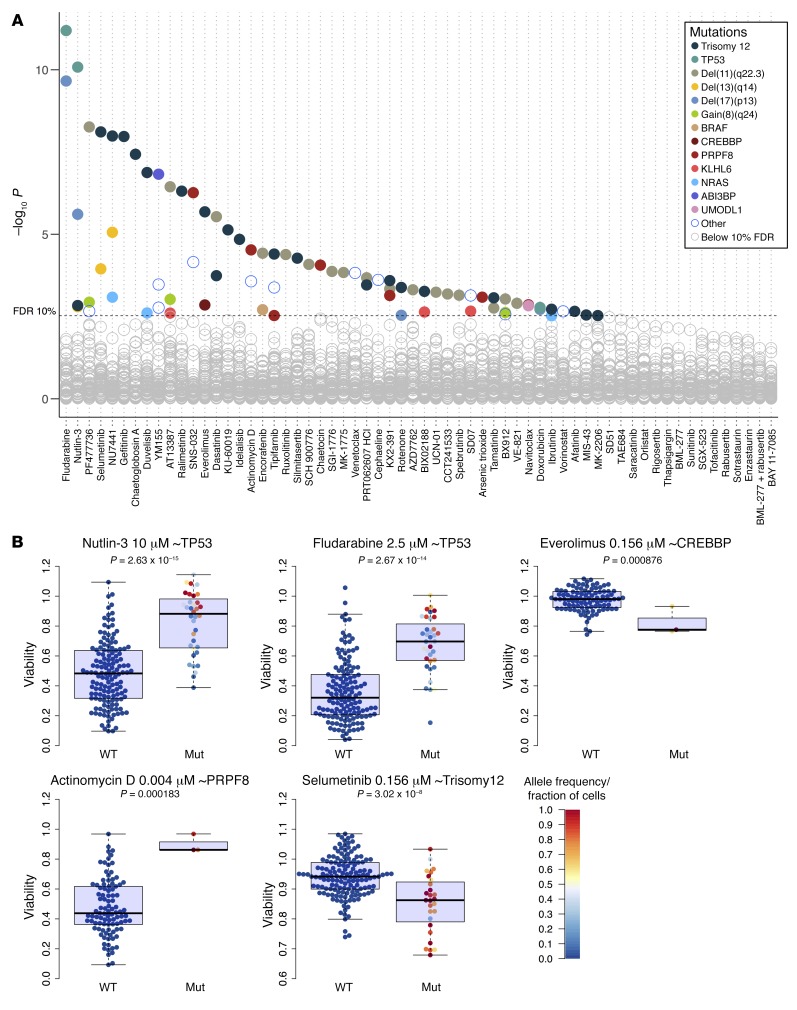
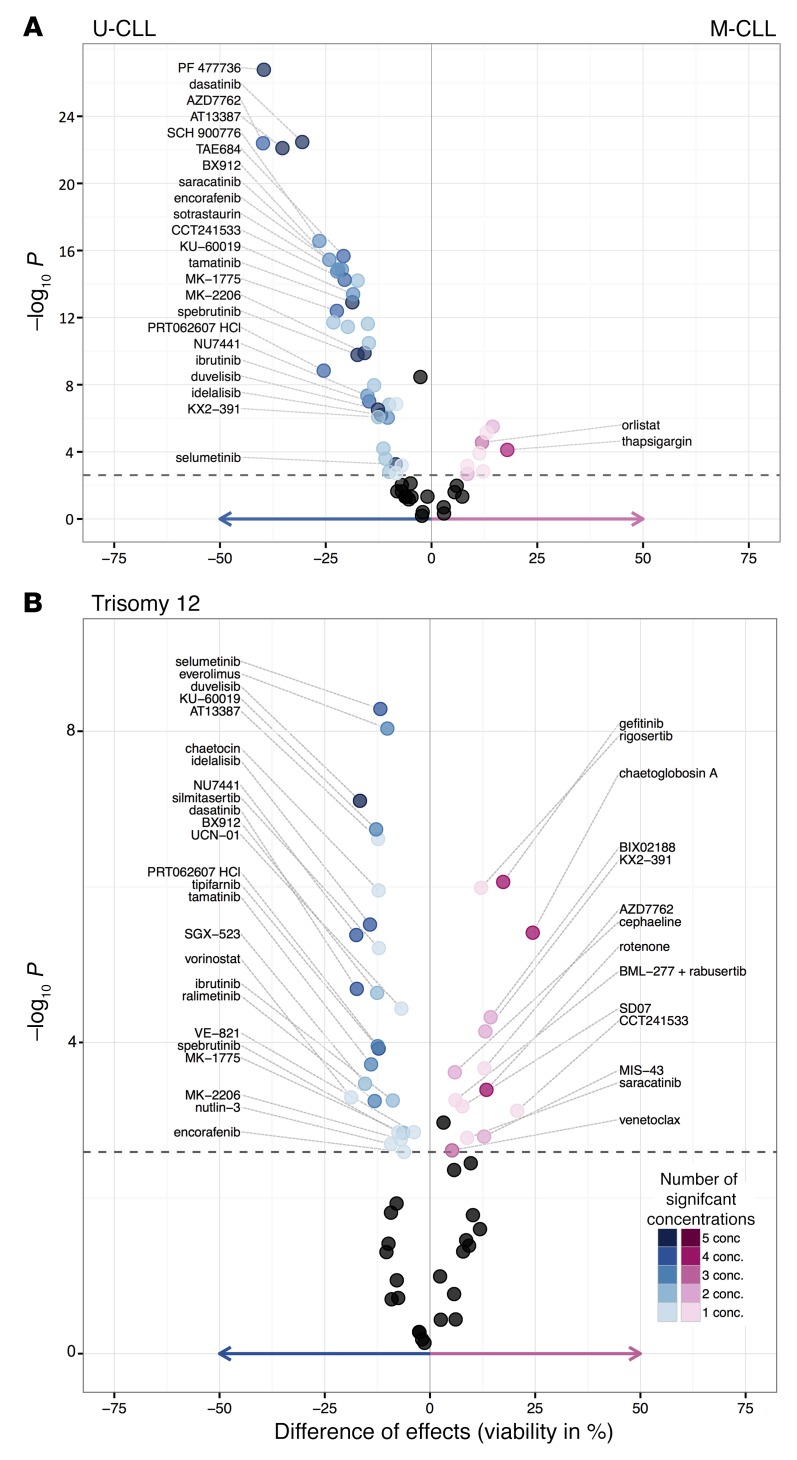
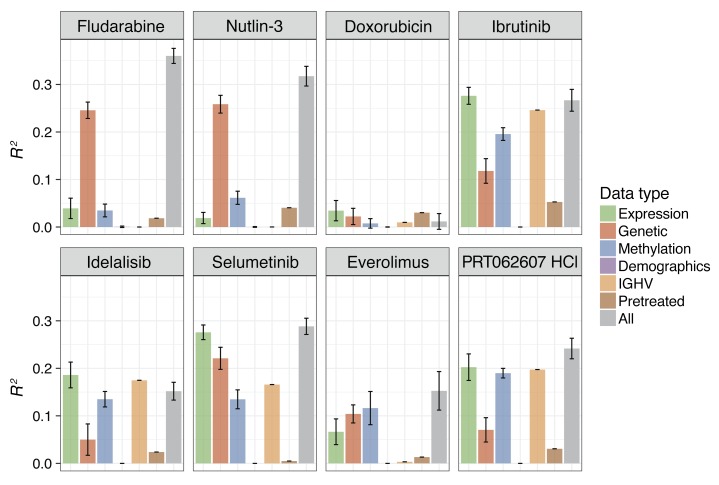
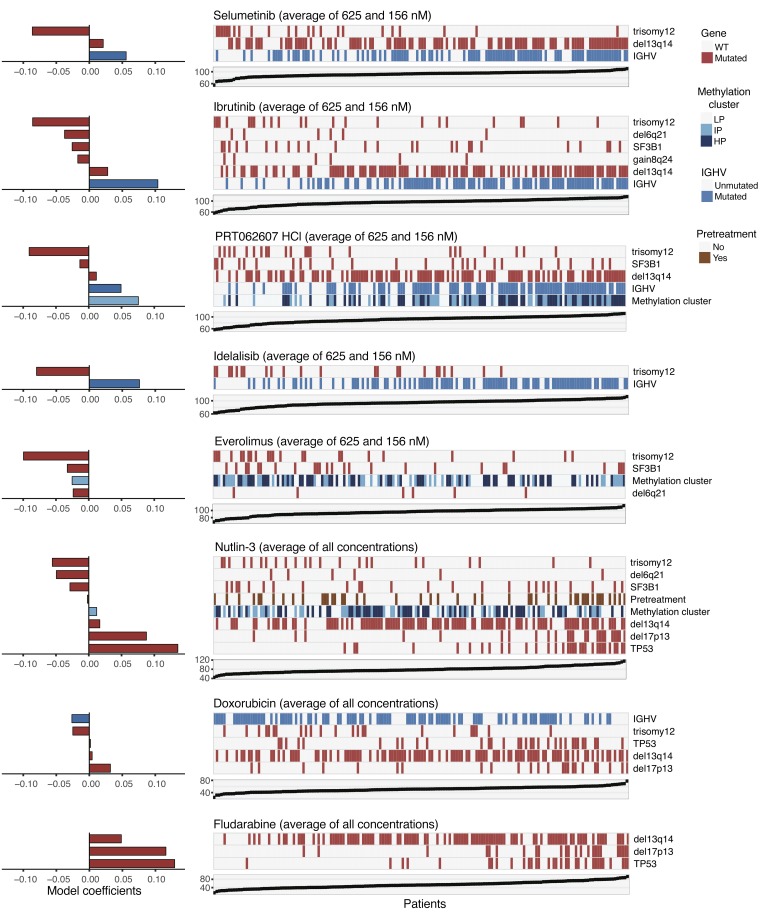
References
-
- de Gramont A, et al. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nat Rev Clin Oncol. 2015;12(4):197–212. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
