

Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain
- PMID: 29227469
- PMCID: PMC5951394
- DOI: 10.1038/nbt.4038
Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain
Abstract
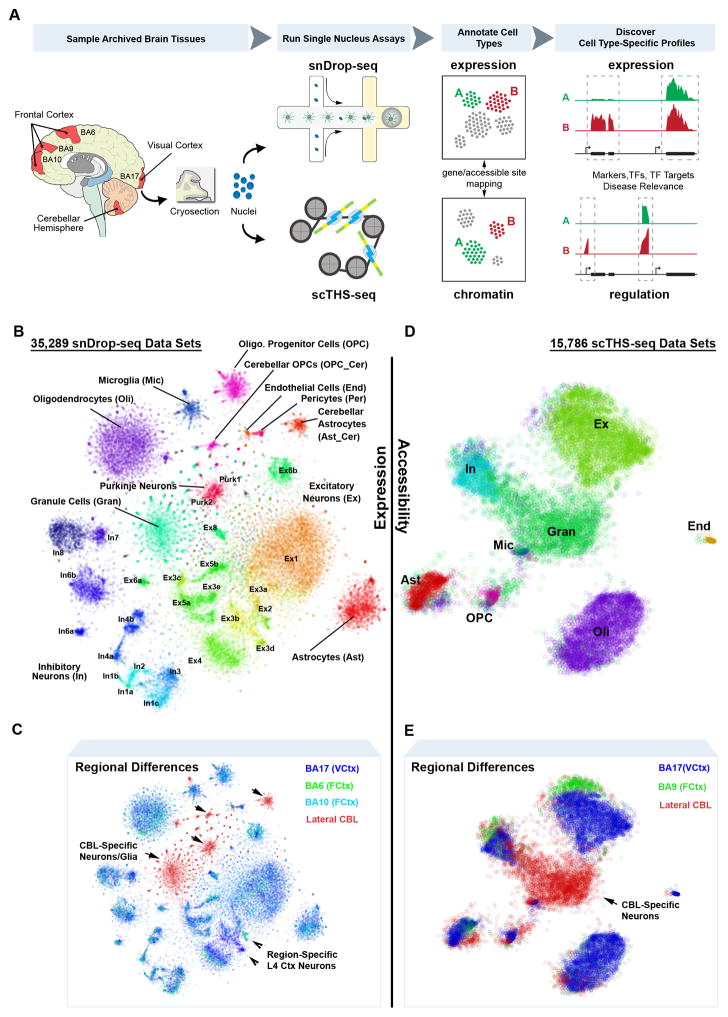
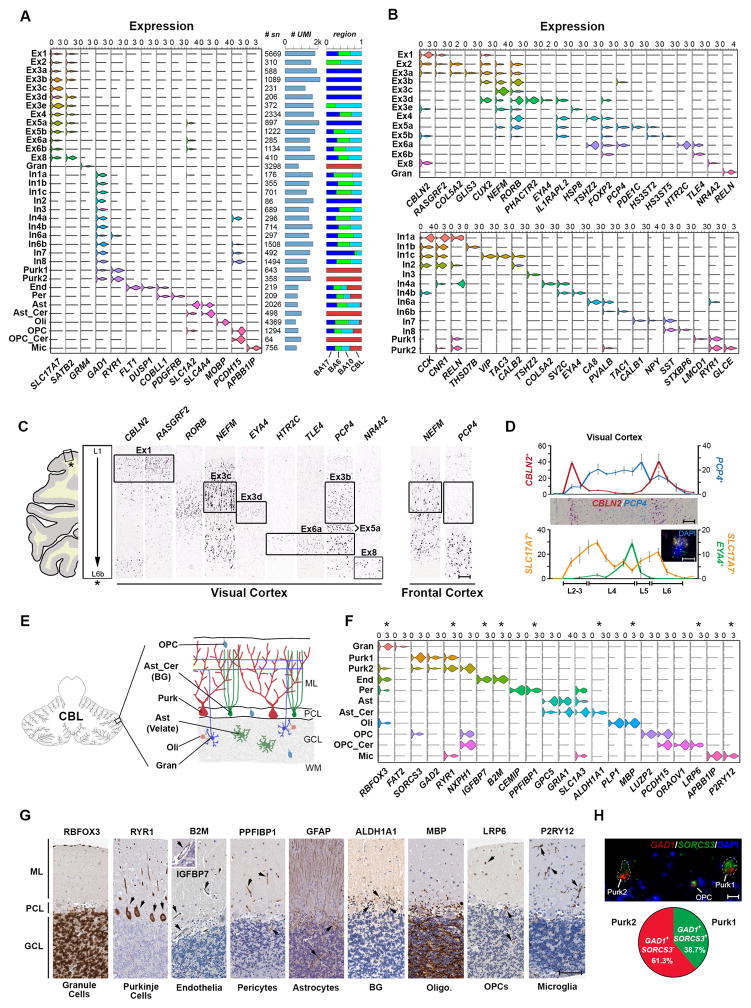
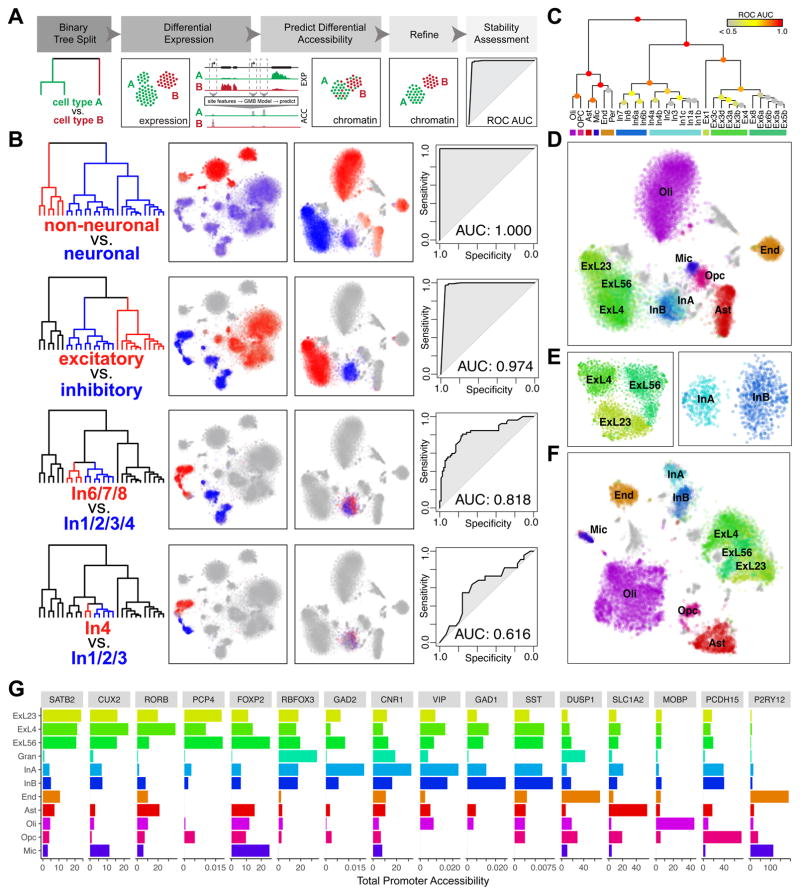
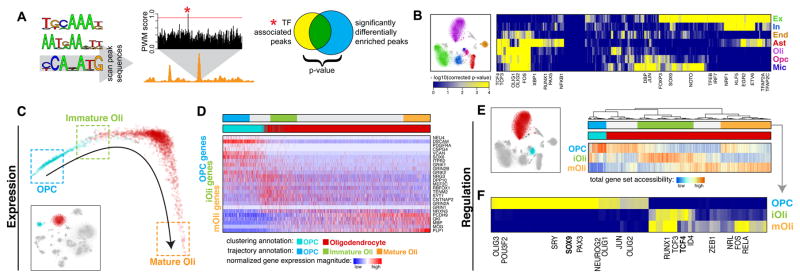
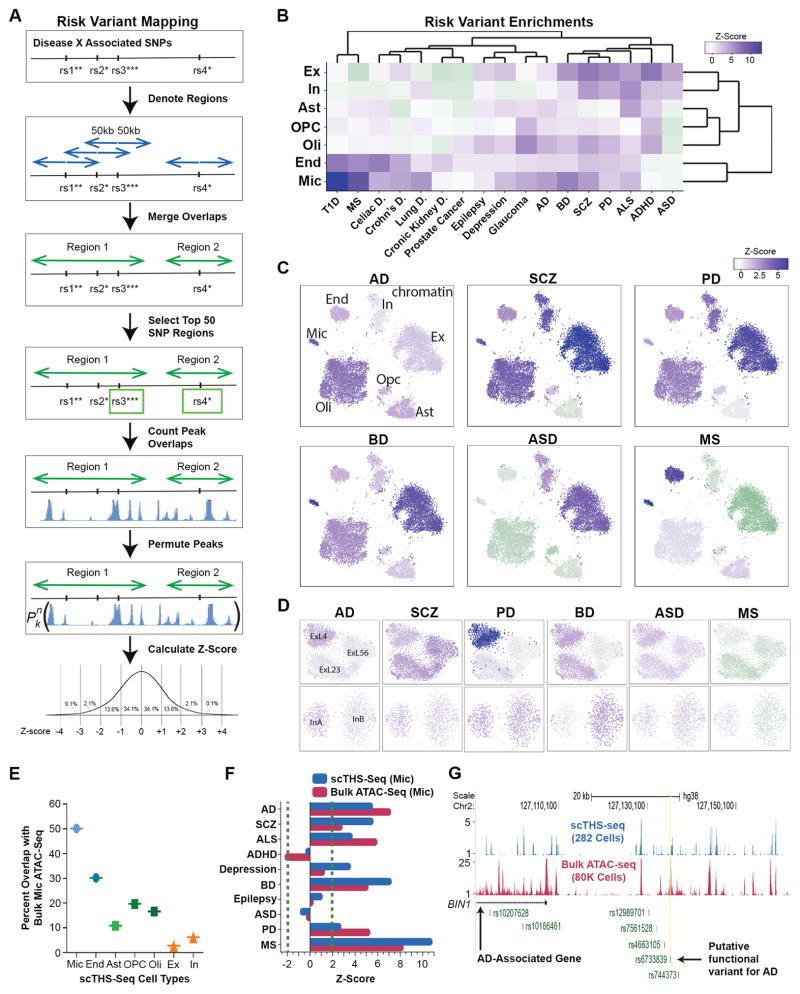
Detailed characterization of the cell types in the human brain requires scalable experimental approaches to examine multiple aspects of the molecular state of individual cells, as well as computational integration of the data to produce unified cell-state annotations. Here we report improved high-throughput methods for single-nucleus droplet-based sequencing (snDrop-seq) and single-cell transposome hypersensitive site sequencing (scTHS-seq). We used each method to acquire nuclear transcriptomic and DNA accessibility maps for >60,000 single cells from human adult visual cortex, frontal cortex, and cerebellum. Integration of these data revealed regulatory elements and transcription factors that underlie cell-type distinctions, providing a basis for the study of complex processes in the brain, such as genetic programs that coordinate adult remyelination. We also mapped disease-associated risk variants to specific cellular populations, which provided insights into normal and pathogenic cellular processes in the human brain. This integrative multi-omics approach permits more detailed single-cell interrogation of complex organs and tissues.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Comment in
-
Strength in numbers from integrated single-cell neuroscience.Nat Biotechnol. 2018 Jan 10;36(1):41-42. doi: 10.1038/nbt.4055. Nat Biotechnol. 2018. PMID: 29319686 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
