

Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs
- PMID: 29227473
- PMCID: PMC5771956
- DOI: 10.1038/nchembio.2527
Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs
Abstract
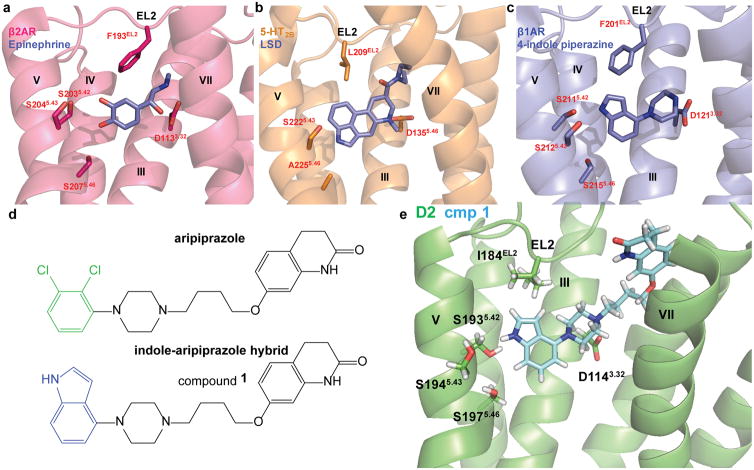
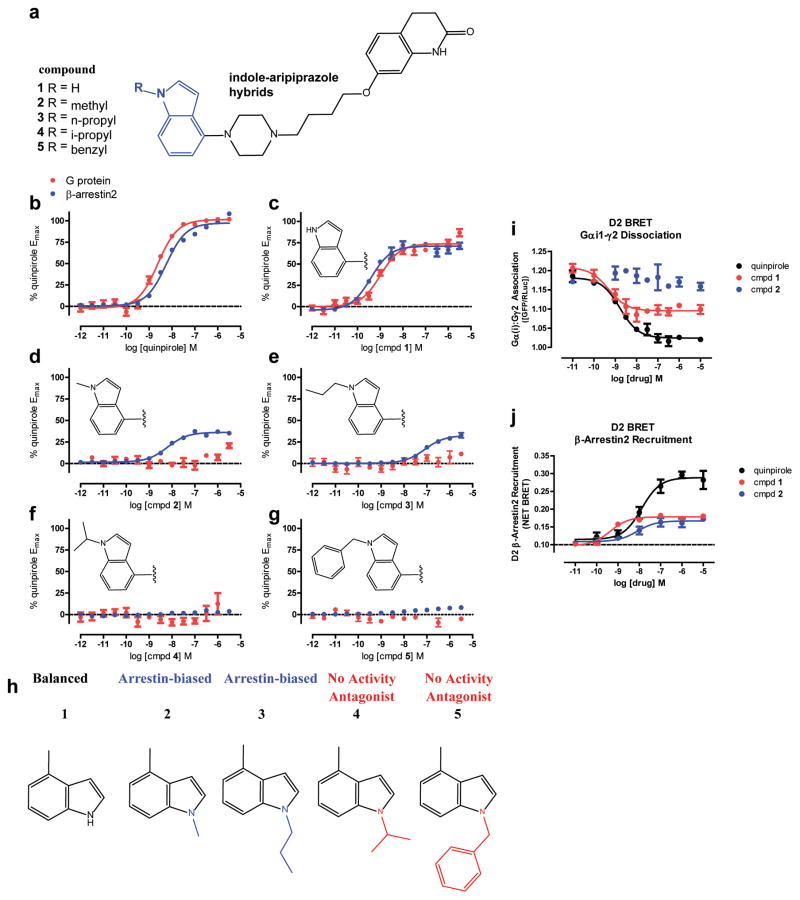
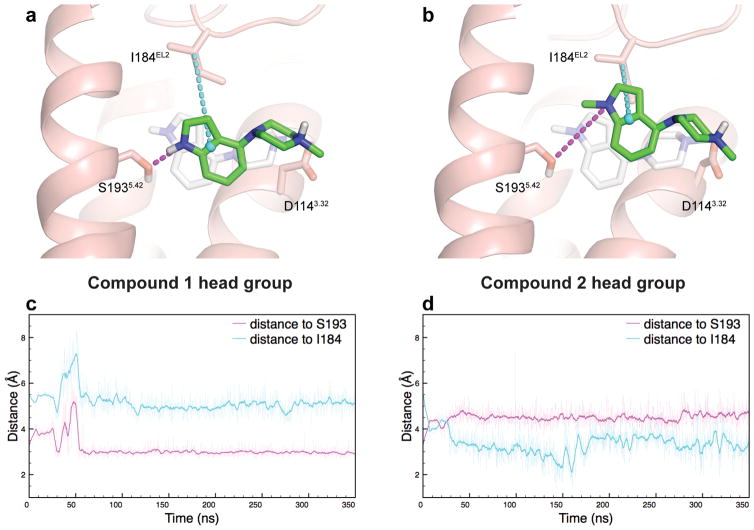
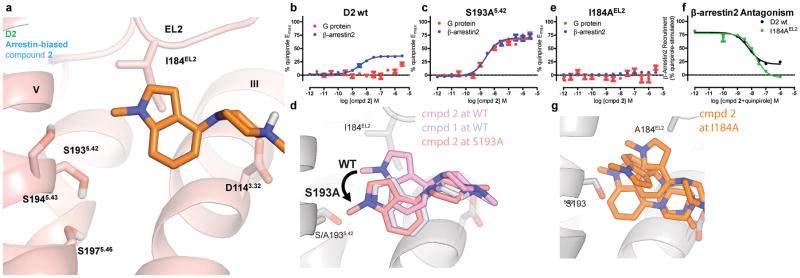
Development of biased ligands targeting G protein-coupled receptors (GPCRs) is a promising approach for current drug discovery. Although structure-based drug design of biased agonists remains challenging even with an abundance of GPCR crystal structures, we present an approach for translating GPCR structural data into β-arrestin-biased ligands for aminergic GPCRs. We identified specific amino acid-ligand contacts at transmembrane helix 5 (TM5) and extracellular loop 2 (EL2) responsible for Gi/o and β-arrestin signaling, respectively, and targeted those residues to develop biased ligands. For these ligands, we found that bias is conserved at other aminergic GPCRs that retain similar residues at TM5 and EL2. Our approach provides a template for generating arrestin-biased ligands by modifying predicted ligand interactions that block TM5 interactions and promote EL2 interactions. This strategy may facilitate the structure-guided design of arrestin-biased ligands at other GPCRs, including polypharmacological biased ligands.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
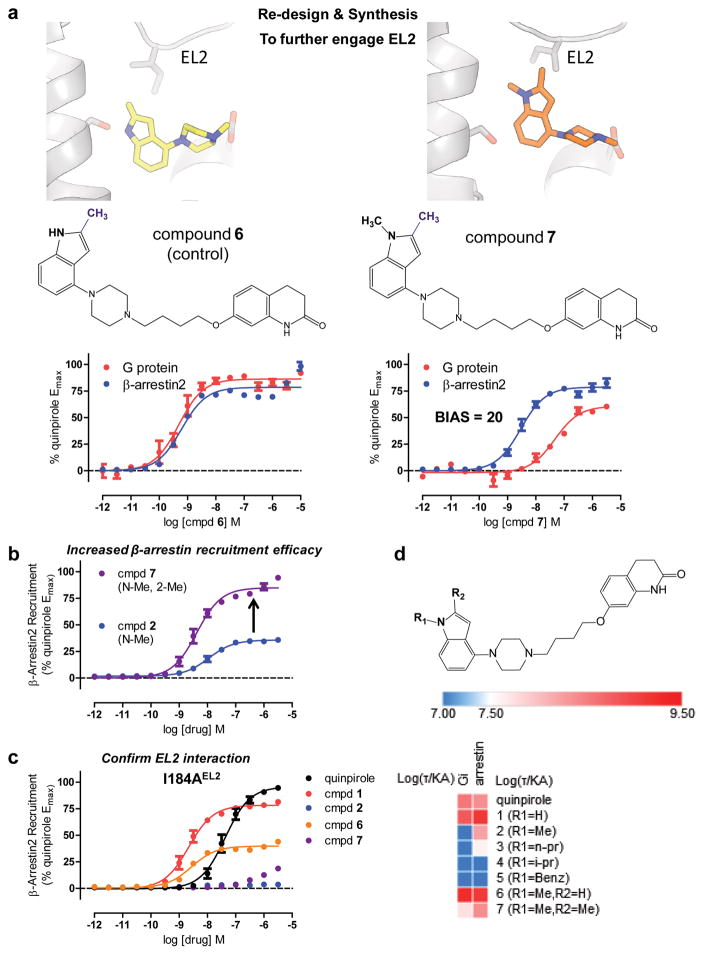
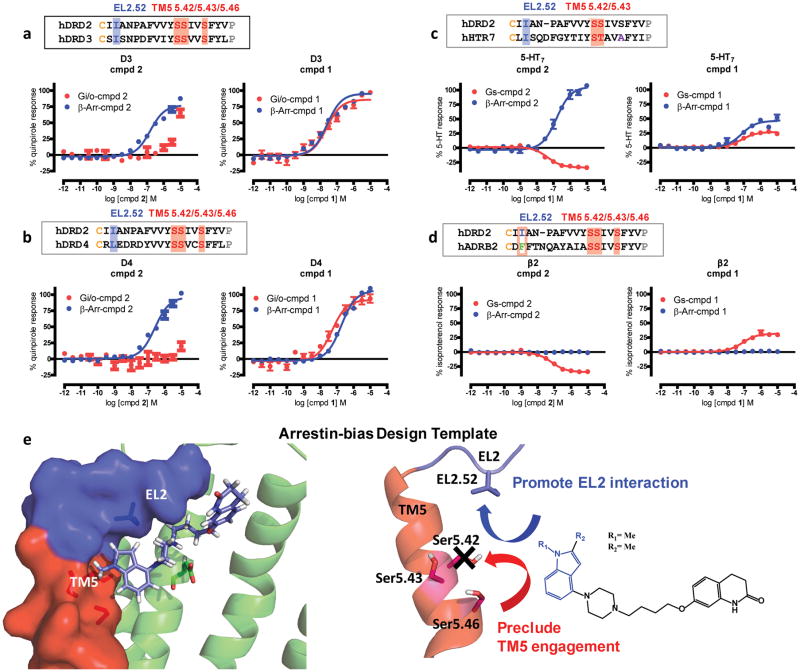
References
-
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. - PubMed
-
- Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. - PubMed
-
- Urban JD, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. - PubMed
-
- Dewire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. - PubMed
Publication types
MeSH terms
Substances
Associated data
- PubChem-Substance/348353604
- PubChem-Substance/348353607
- PubChem-Substance/348353608
- PubChem-Substance/348353609
- PubChem-Substance/348353610
- PubChem-Substance/348353611
- PubChem-Substance/348353612
- PubChem-Substance/348353613
- PubChem-Substance/348353614
- PubChem-Substance/348353605
- PubChem-Substance/348353606
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
