

KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer
- PMID: 29229669
- PMCID: PMC5995645
- DOI: 10.1101/cshperspect.a031435
KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer
Abstract
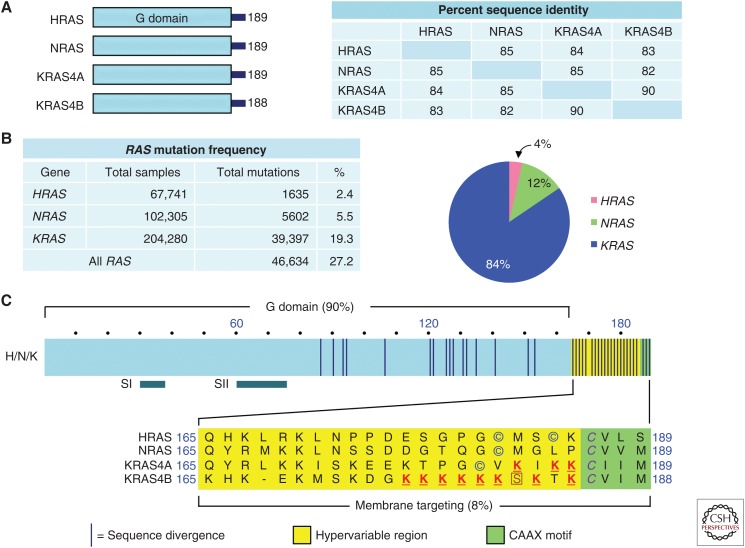
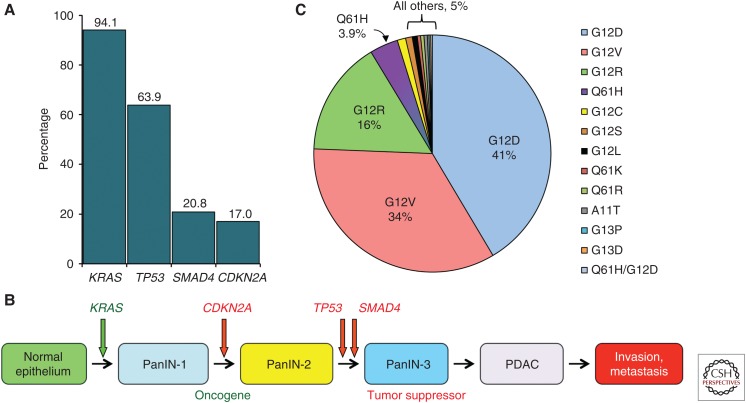
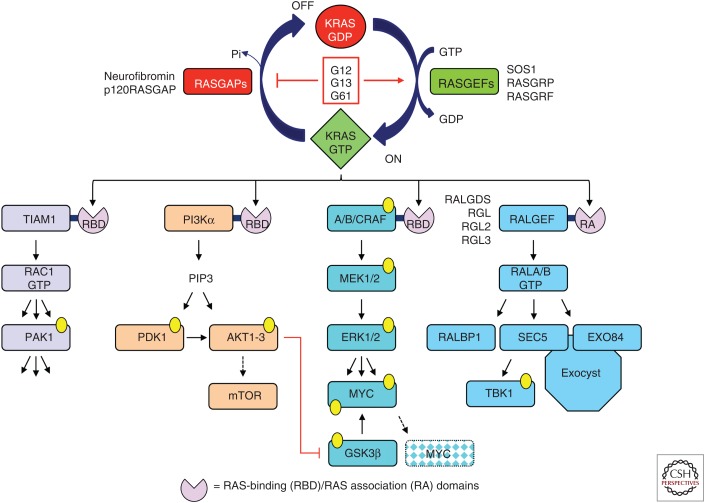
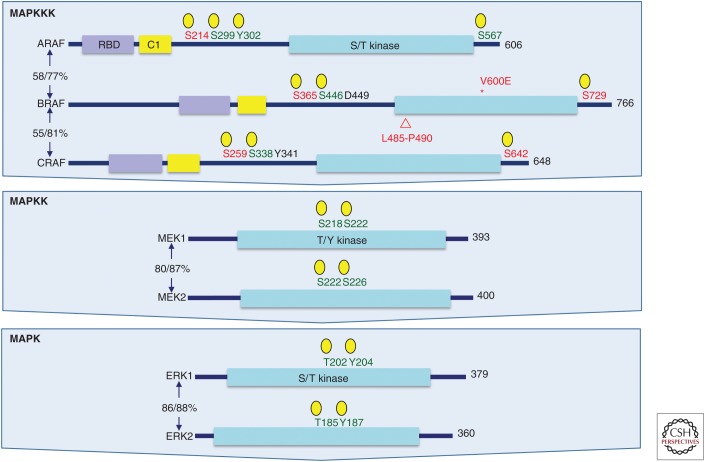
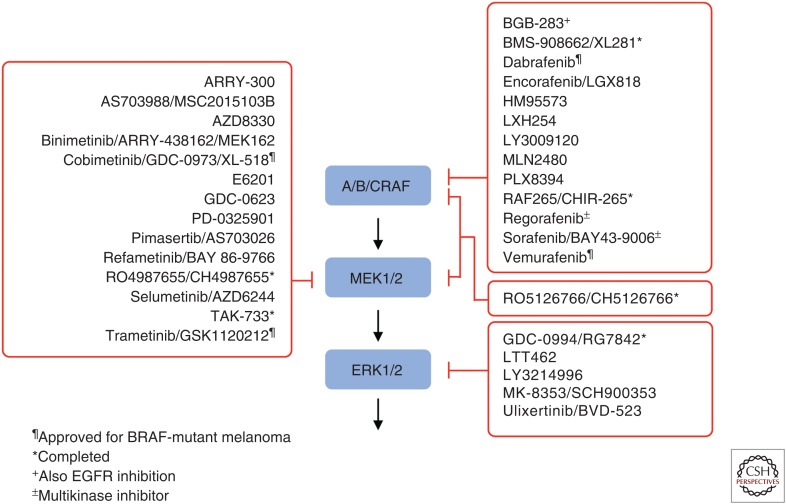
RAS genes (HRAS, KRAS, and NRAS) comprise the most frequently mutated oncogene family in human cancer. With the highest RAS mutation frequencies seen with the top three causes of cancer deaths in the United States (lung, colorectal, and pancreatic cancer), the development of anti-RAS therapies is a major priority for cancer research. Despite more than three decades of intense effort, no effective RAS inhibitors have yet to reach the cancer patient. With bitter lessons learned from past failures and with new ideas and strategies, there is renewed hope that undruggable RAS may finally be conquered. With the KRAS isoform mutated in 84% of all RAS-mutant cancers, we focus on KRAS. With a near 100% KRAS mutation frequency, pancreatic ductal adenocarcinoma (PDAC) is considered the most RAS-addicted of all cancers. We review the role of KRAS as a driver and therapeutic target in PDAC.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. 1988. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53: 549–554. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous