

Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium
- PMID: 29229834
- PMCID: PMC5748227
- DOI: 10.1073/pnas.1717891115
Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium
Abstract
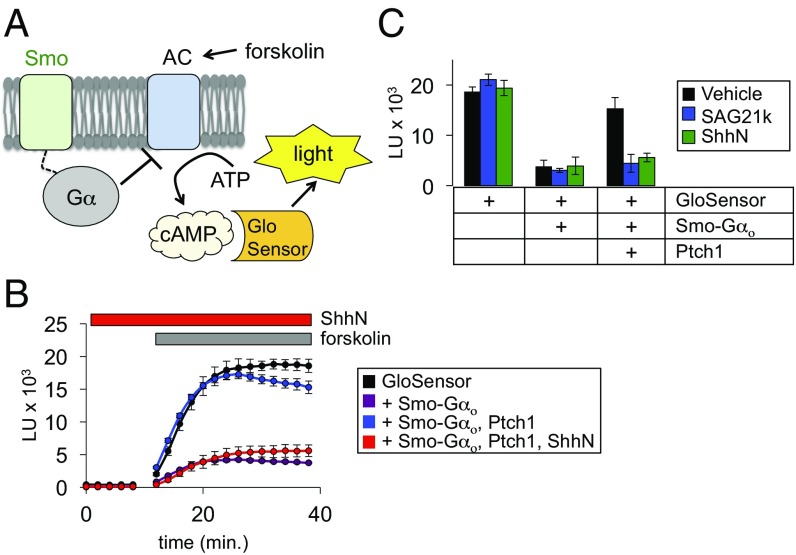
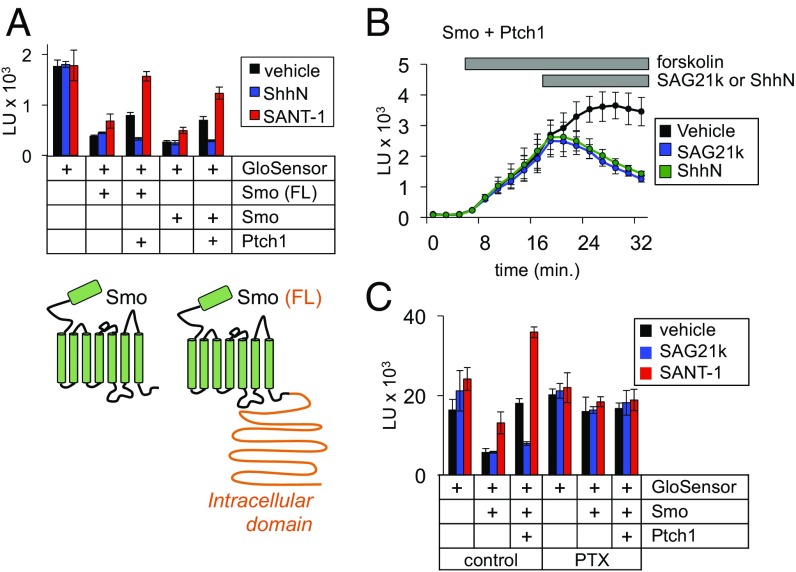
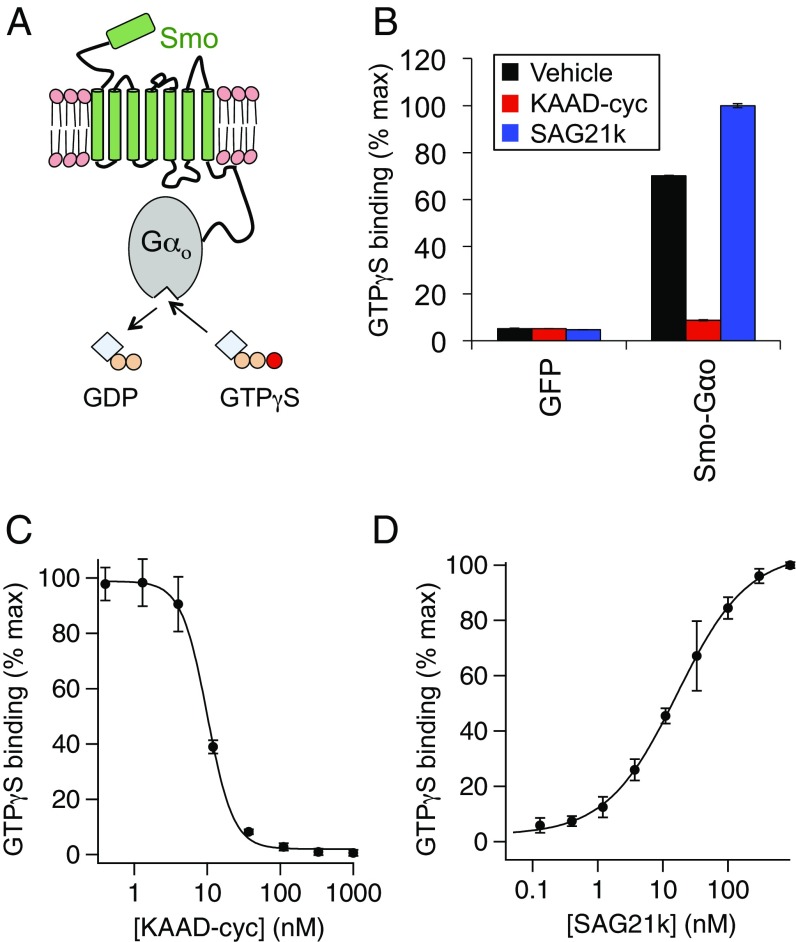
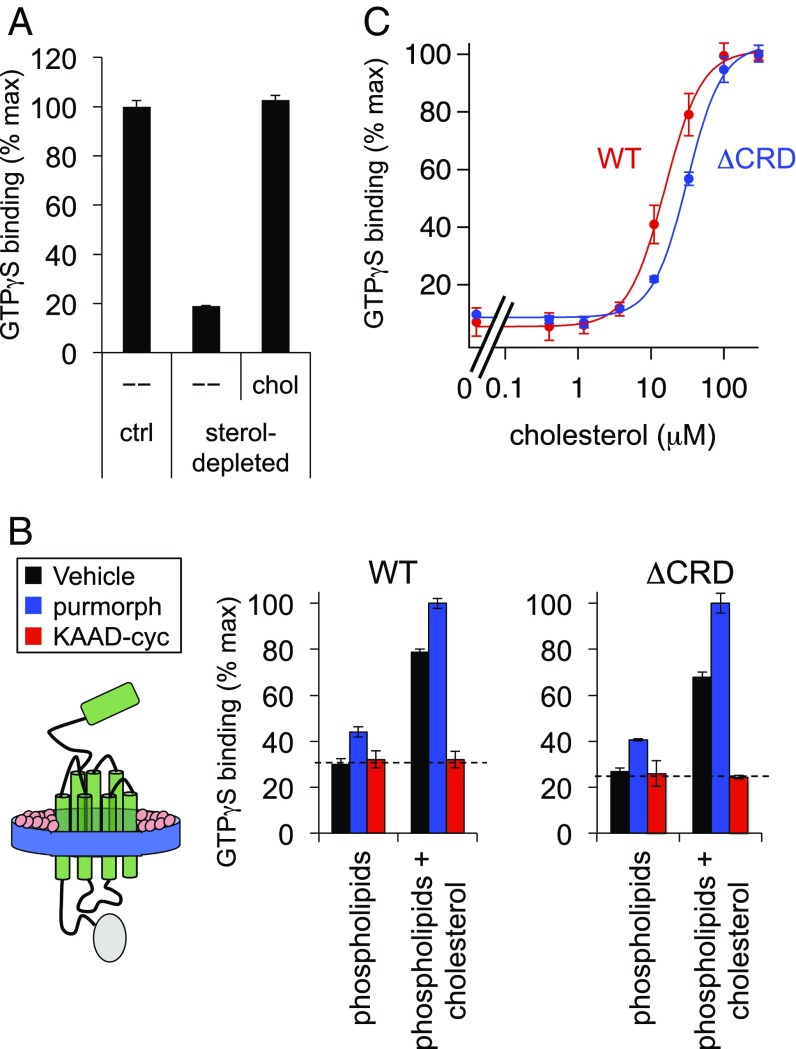
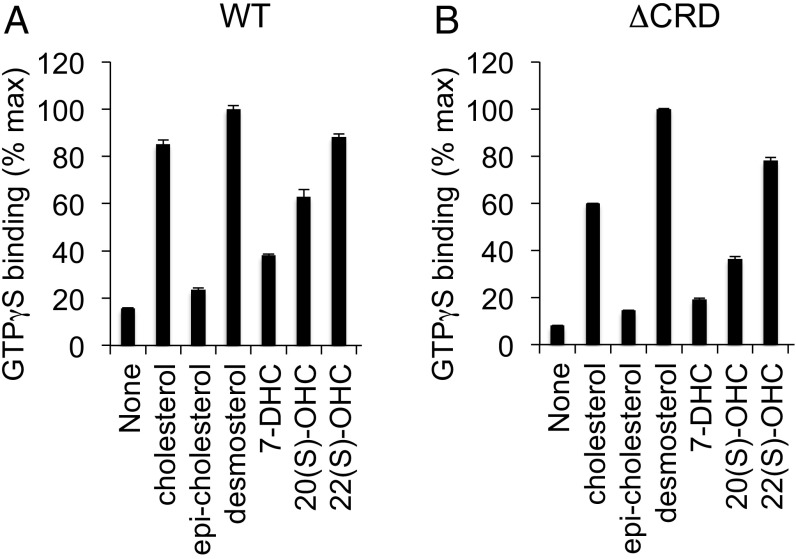
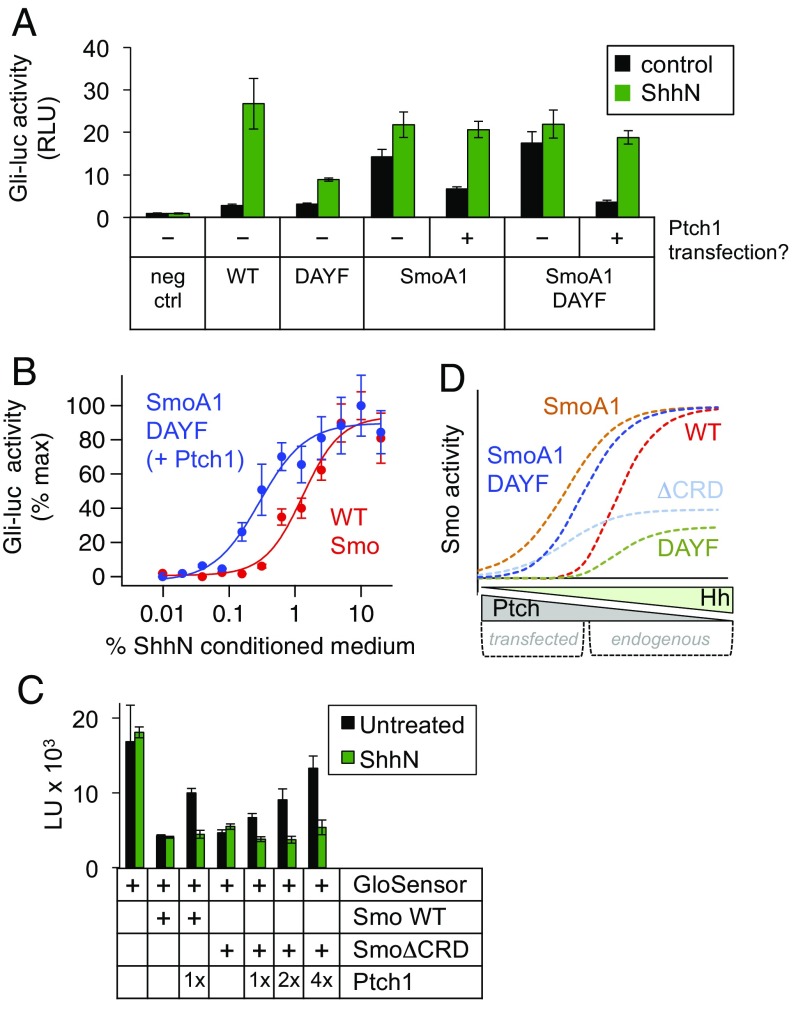
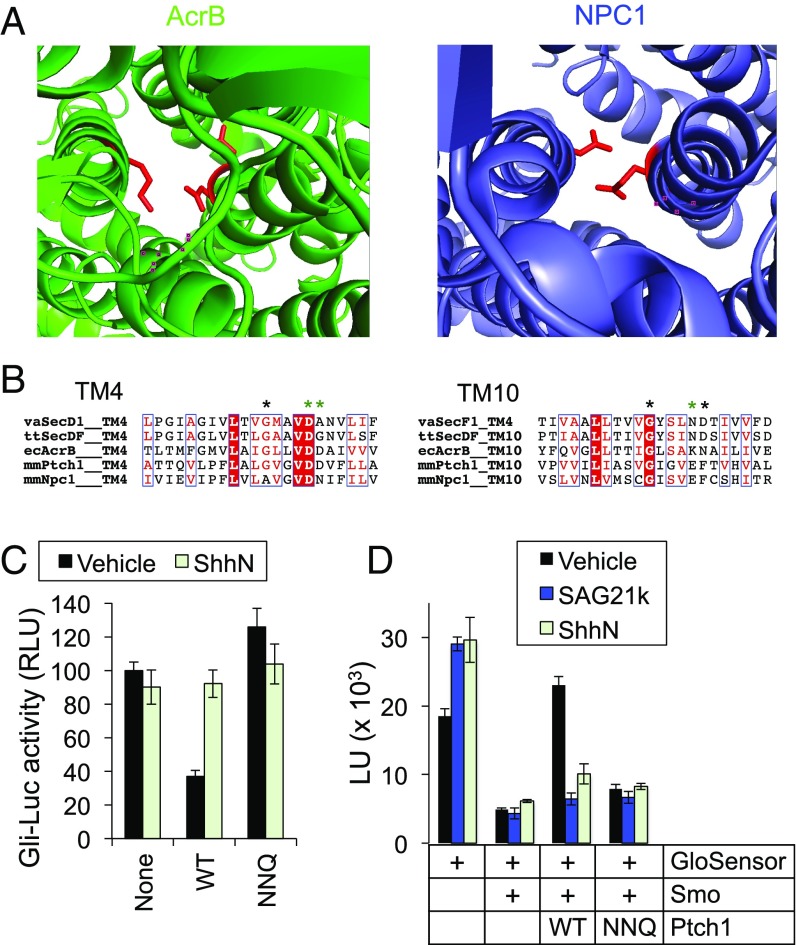
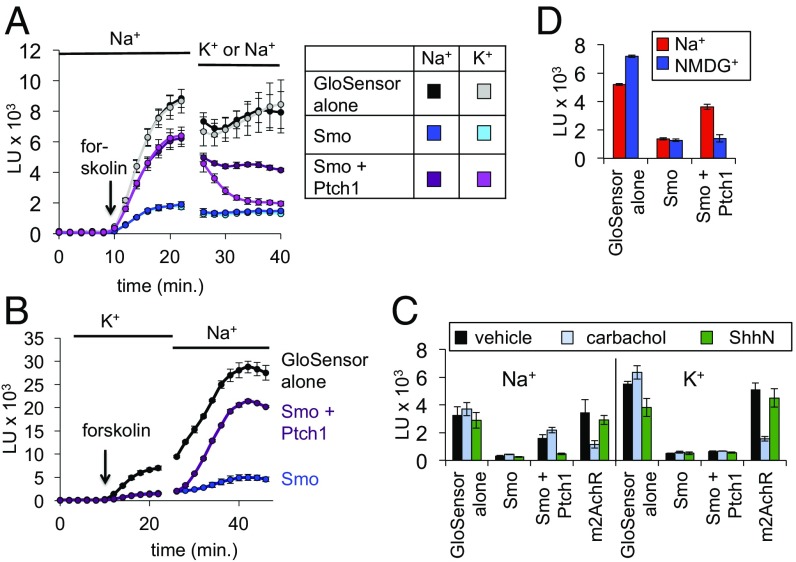
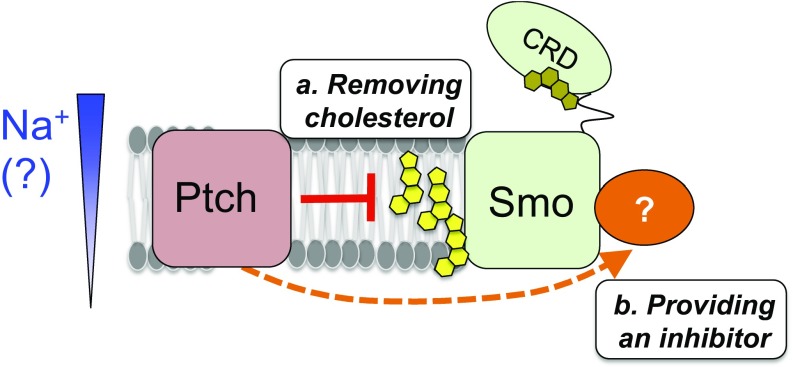
Hedgehog signaling specifies tissue patterning and renewal, and pathway components are commonly mutated in certain malignancies. Although central to ensuring appropriate pathway activity in all Hedgehog-responsive cells, how the transporter-like receptor Patched1 regulates the seven-transmembrane protein Smoothened remains mysterious, partially due to limitations in existing tools and experimental systems. Here we employ direct, real-time, biochemical and physiology-based approaches to monitor Smoothened activity in cellular and in vitro contexts. Patched1-Smoothened coupling is rapid, dynamic, and can be recapitulated without cilium-specific proteins or lipids. By reconstituting purified Smoothened in vitro, we show that cholesterol within the bilayer is sufficient for constitutive Smoothened activation. Cholesterol effects occur independently of the lipid-binding Smoothened extracellular domain, a region that is dispensable for Patched1-Smoothened coupling. Finally, we show that Patched1 specifically requires extracellular Na+ to regulate Smoothened in our assays, raising the possibility that a Na+ gradient provides the energy source for Patched1 catalytic activity. Our work suggests a hypothesis wherein Patched1, chemiosmotically driven by the transmembrane Na+ gradient common to metazoans, regulates Smoothened by shielding its heptahelical domain from cholesterol, or by providing an inhibitor that overrides this cholesterol activation.
Keywords: Hedgehog; Na+; Patched; Smoothened; cholesterol.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Smoothened Sensor Places Sodium and Sterols Center Stage.Dev Cell. 2018 Jan 8;44(1):3-4. doi: 10.1016/j.devcel.2017.12.015. Epub 2018 Jan 8. Dev Cell. 2018. PMID: 29316438
Similar articles
-
Patched1-ArhGAP36-PKA-Inversin axis determines the ciliary translocation of Smoothened for Sonic Hedgehog pathway activation.Proc Natl Acad Sci U S A. 2019 Jan 15;116(3):874-879. doi: 10.1073/pnas.1804042116. Epub 2018 Dec 31. Proc Natl Acad Sci U S A. 2019. PMID: 30598432 Free PMC article.
-
Mechanism and ultrasensitivity in Hedgehog signaling revealed by Patched1 disease mutations.Proc Natl Acad Sci U S A. 2021 Feb 9;118(6):e2006800118. doi: 10.1073/pnas.2006800118. Proc Natl Acad Sci U S A. 2021. PMID: 33526656 Free PMC article.
-
Distinct Cation Gradients Power Cholesterol Transport at Different Key Points in the Hedgehog Signaling Pathway.Dev Cell. 2020 Nov 9;55(3):314-327.e7. doi: 10.1016/j.devcel.2020.08.002. Epub 2020 Aug 28. Dev Cell. 2020. PMID: 32860743 Free PMC article.
-
Cholesterol access in cellular membranes controls Hedgehog signaling.Nat Chem Biol. 2020 Dec;16(12):1303-1313. doi: 10.1038/s41589-020-00678-2. Epub 2020 Nov 16. Nat Chem Biol. 2020. PMID: 33199907 Free PMC article. Review.
-
Structures of vertebrate Patched and Smoothened reveal intimate links between cholesterol and Hedgehog signalling.Curr Opin Struct Biol. 2019 Aug;57:204-214. doi: 10.1016/j.sbi.2019.05.015. Epub 2019 Jun 24. Curr Opin Struct Biol. 2019. PMID: 31247512 Free PMC article. Review.
Cited by
-
Hedgehog-interacting protein acts in the habenula to regulate nicotine intake.Proc Natl Acad Sci U S A. 2022 Nov 15;119(46):e2209870119. doi: 10.1073/pnas.2209870119. Epub 2022 Nov 8. Proc Natl Acad Sci U S A. 2022. PMID: 36346845 Free PMC article.
-
Hedgehog signaling promotes lipolysis in adipose tissue through directly regulating Bmm/ATGL lipase.Dev Biol. 2020 Jan 1;457(1):128-139. doi: 10.1016/j.ydbio.2019.09.009. Epub 2019 Sep 21. Dev Biol. 2020. PMID: 31550483 Free PMC article.
-
Patched1-ArhGAP36-PKA-Inversin axis determines the ciliary translocation of Smoothened for Sonic Hedgehog pathway activation.Proc Natl Acad Sci U S A. 2019 Jan 15;116(3):874-879. doi: 10.1073/pnas.1804042116. Epub 2018 Dec 31. Proc Natl Acad Sci U S A. 2019. PMID: 30598432 Free PMC article.
-
Cholesterol and Hedgehog Signaling: Mutual Regulation and Beyond.Front Cell Dev Biol. 2022 Apr 27;10:774291. doi: 10.3389/fcell.2022.774291. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35573688 Free PMC article. Review.
-
Smoothened (SMO) regulates insulin-like growth factor 1 receptor (IGF1R) levels and protein kinase B (AKT) localization and signaling.Lab Invest. 2022 Apr;102(4):401-410. doi: 10.1038/s41374-021-00702-6. Epub 2021 Dec 10. Lab Invest. 2022. PMID: 34893758 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
