

Structure-based prediction of ligand-protein interactions on a genome-wide scale
- PMID: 29229851
- PMCID: PMC5748165
- DOI: 10.1073/pnas.1705381114
Structure-based prediction of ligand-protein interactions on a genome-wide scale
Abstract
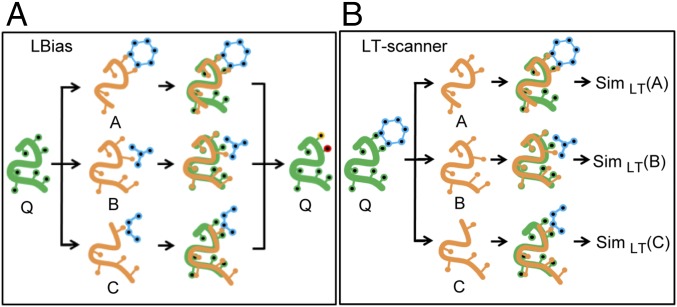
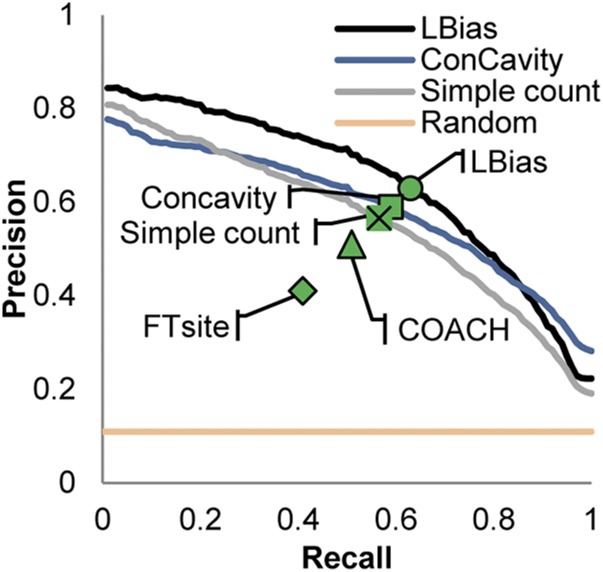
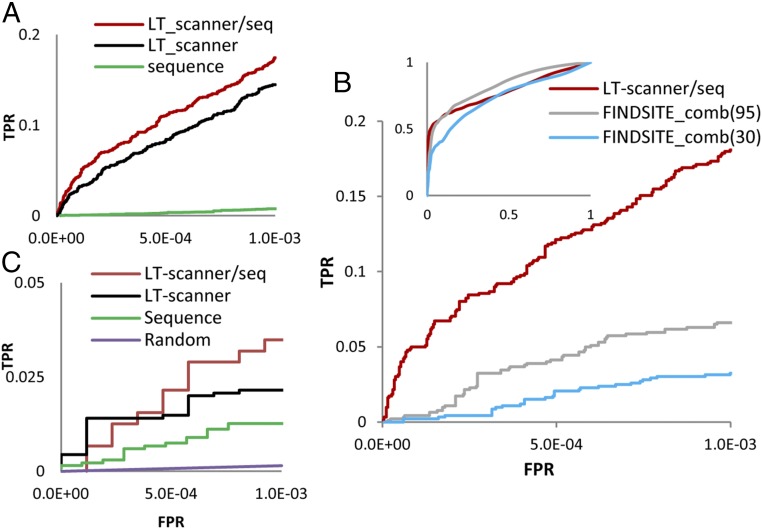
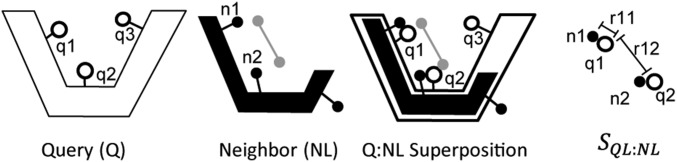
We report a template-based method, LT-scanner, which scans the human proteome using protein structural alignment to identify proteins that are likely to bind ligands that are present in experimentally determined complexes. A scoring function that rapidly accounts for binding site similarities between the template and the proteins being scanned is a crucial feature of the method. The overall approach is first tested based on its ability to predict the residues on the surface of a protein that are likely to bind small-molecule ligands. The algorithm that we present, LBias, is shown to compare very favorably to existing algorithms for binding site residue prediction. LT-scanner's performance is evaluated based on its ability to identify known targets of Food and Drug Administration (FDA)-approved drugs and it too proves to be highly effective. The specificity of the scoring function that we use is demonstrated by the ability of LT-scanner to identify the known targets of FDA-approved kinase inhibitors based on templates involving other kinases. Combining sequence with structural information further improves LT-scanner performance. The approach we describe is extendable to the more general problem of identifying binding partners of known ligands even if they do not appear in a structurally determined complex, although this will require the integration of methods that combine protein structure and chemical compound databases.
Keywords: drug off-targets; machine learning; protein–ligand interactions; structure-based prediction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
DNABind: a hybrid algorithm for structure-based prediction of DNA-binding residues by combining machine learning- and template-based approaches.Proteins. 2013 Nov;81(11):1885-99. doi: 10.1002/prot.24330. Epub 2013 Aug 16. Proteins. 2013. PMID: 23737141
-
Domain-based small molecule binding site annotation.BMC Bioinformatics. 2006 Mar 17;7:152. doi: 10.1186/1471-2105-7-152. BMC Bioinformatics. 2006. PMID: 16545112 Free PMC article.
-
Nonlinear scoring functions for similarity-based ligand docking and binding affinity prediction.J Chem Inf Model. 2013 Nov 25;53(11):3097-112. doi: 10.1021/ci400510e. Epub 2013 Nov 11. J Chem Inf Model. 2013. PMID: 24171431
-
Polypharmacology rescored: protein-ligand interaction profiles for remote binding site similarity assessment.Prog Biophys Mol Biol. 2014 Nov-Dec;116(2-3):174-86. doi: 10.1016/j.pbiomolbio.2014.05.006. Epub 2014 Jun 9. Prog Biophys Mol Biol. 2014. PMID: 24923864 Review.
-
Prediction of protein-protein interaction based on structure.Methods Mol Biol. 2006;340:207-34. doi: 10.1385/1-59745-116-9:207. Methods Mol Biol. 2006. PMID: 16957339 Review.
Cited by
-
Protein functional annotation of simultaneously improved stability, accuracy and false discovery rate achieved by a sequence-based deep learning.Brief Bioinform. 2020 Jul 15;21(4):1437-1447. doi: 10.1093/bib/bbz081. Brief Bioinform. 2020. PMID: 31504150 Free PMC article.
-
Computational insights of phytochemical-driven disruption of RNA-dependent RNA polymerase-mediated replication of coronavirus: a strategic treatment plan against coronavirus disease 2019.New Microbes New Infect. 2021 May;41:100878. doi: 10.1016/j.nmni.2021.100878. Epub 2021 Mar 31. New Microbes New Infect. 2021. PMID: 33815808 Free PMC article.
-
ClusPro LigTBM: Automated Template-based Small Molecule Docking.J Mol Biol. 2020 May 15;432(11):3404-3410. doi: 10.1016/j.jmb.2019.12.011. Epub 2019 Dec 19. J Mol Biol. 2020. PMID: 31863748 Free PMC article.
-
Computational methods and tools for binding site recognition between proteins and small molecules: from classical geometrical approaches to modern machine learning strategies.J Comput Aided Mol Des. 2019 Oct;33(10):887-903. doi: 10.1007/s10822-019-00235-7. Epub 2019 Oct 18. J Comput Aided Mol Des. 2019. PMID: 31628659
-
Strategy for the Biosynthesis of Short Oligopeptides: Green and Sustainable Chemistry.Biomolecules. 2019 Nov 13;9(11):733. doi: 10.3390/biom9110733. Biomolecules. 2019. PMID: 31766233 Free PMC article. Review.
References
-
- Hendlich M, Rippmann F, Barnickel G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–363, 389. - PubMed
-
- Laskowski RA. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph. 1995;13:323–330. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
