

Annotation-free quantification of RNA splicing using LeafCutter
- PMID: 29229983
- PMCID: PMC5742080
- DOI: 10.1038/s41588-017-0004-9
Annotation-free quantification of RNA splicing using LeafCutter
Abstract
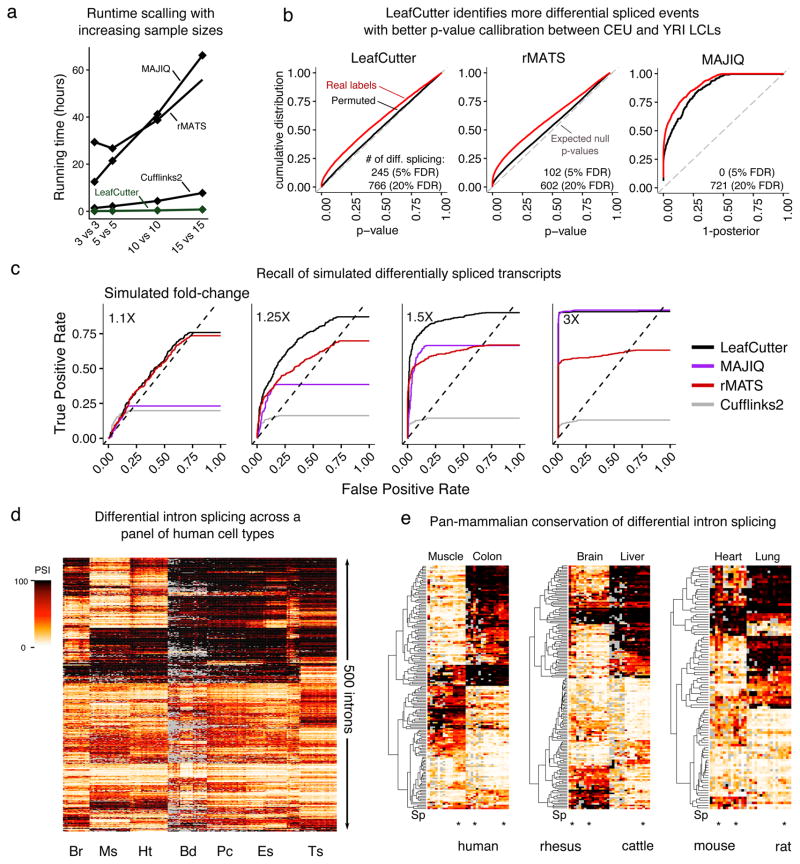
The excision of introns from pre-mRNA is an essential step in mRNA processing. We developed LeafCutter to study sample and population variation in intron splicing. LeafCutter identifies variable splicing events from short-read RNA-seq data and finds events of high complexity. Our approach obviates the need for transcript annotations and circumvents the challenges in estimating relative isoform or exon usage in complex splicing events. LeafCutter can be used both to detect differential splicing between sample groups and to map splicing quantitative trait loci (sQTLs). Compared with contemporary methods, our approach identified 1.4-2.1 times more sQTLs, many of which helped us ascribe molecular effects to disease-associated variants. Transcriptome-wide associations between LeafCutter intron quantifications and 40 complex traits increased the number of associated disease genes at a 5% false discovery rate by an average of 2.1-fold compared with that detected through the use of gene expression levels alone. LeafCutter is fast, scalable, easy to use, and available online.
Conflict of interest statement
The authors declare no competing financial interests.
Figures




References
Methods-only References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
