

DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism
- PMID: 29233090
- PMCID: PMC5727985
- DOI: 10.1186/s12864-017-4353-7
DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism
Abstract
Background: DNA methylation at promoters is largely correlated with inhibition of gene expression. However, the role of DNA methylation at enhancers is not fully understood, although a crosstalk with chromatin marks is expected. Actually, there exist contradictory reports about positive and negative correlations between DNA methylation and H3K4me1, a chromatin hallmark of enhancers.
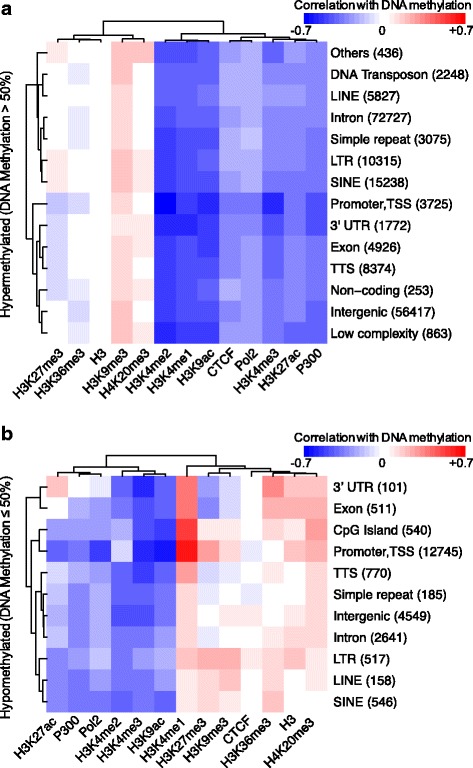
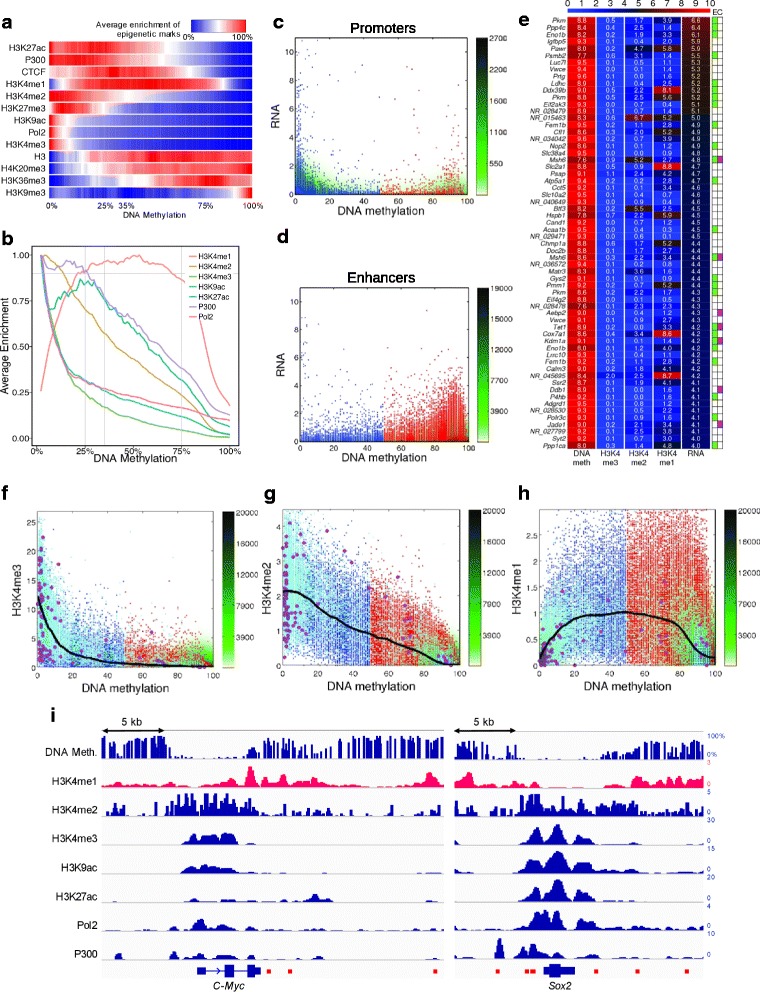
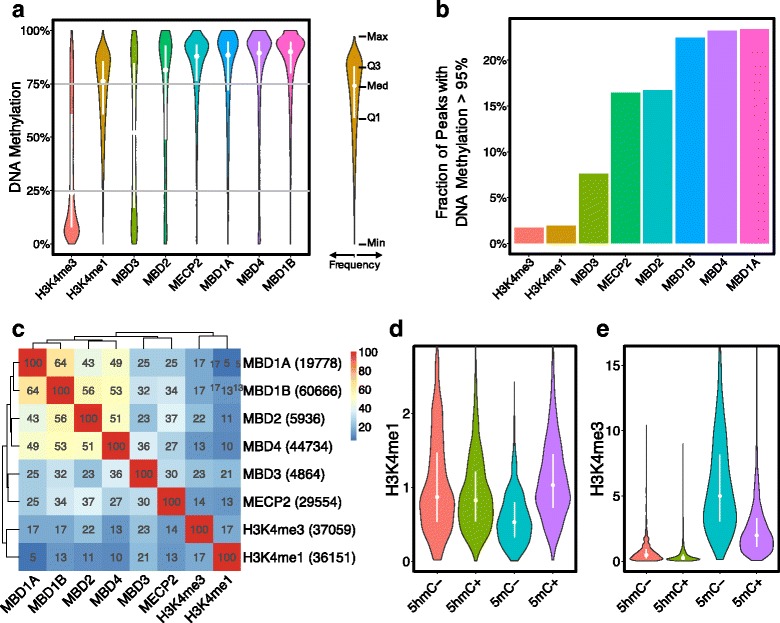
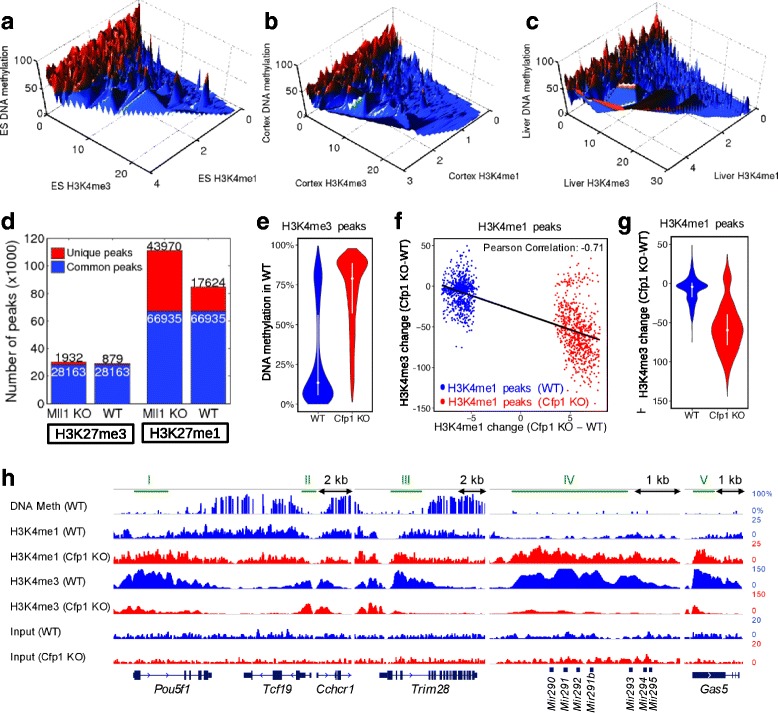
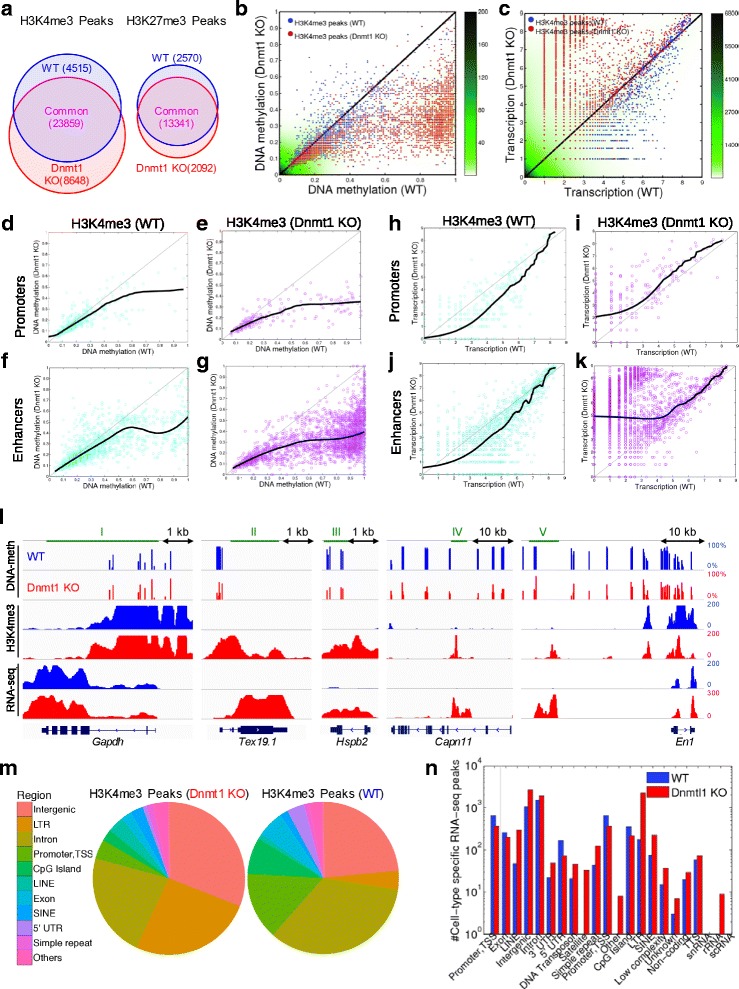
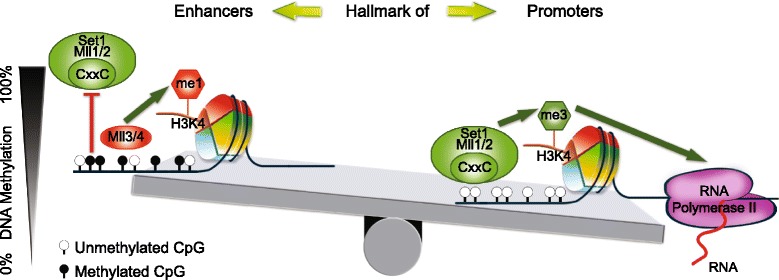
Results: We investigated the relationship between DNA methylation and active chromatin marks through genome-wide correlations, and found anti-correlation between H3K4me1 and H3K4me3 enrichment at low and intermediate DNA methylation loci. We hypothesized "seesaw" dynamics between H3K4me1 and H3K4me3 in the low and intermediate DNA methylation range, in which DNA methylation discriminates between enhancers and promoters, marked by H3K4me1 and H3K4me3, respectively. Low methylated regions are H3K4me3 enriched, while those with intermediate DNA methylation levels are progressively H3K4me1 enriched. Additionally, the enrichment of H3K27ac, distinguishing active from primed enhancers, follows a plateau in the lower range of the intermediate DNA methylation level, corresponding to active enhancers, and decreases linearly in the higher range of the intermediate DNA methylation. Thus, the decrease of the DNA methylation switches smoothly the state of the enhancers from a primed to an active state. We summarize these observations into a rule of thumb of one-out-of-three methylation marks: "In each genomic region only one out of these three methylation marks {DNA methylation, H3K4me1, H3K4me3} is high. If it is the DNA methylation, the region is inactive. If it is H3K4me1, the region is an enhancer, and if it is H3K4me3, the region is a promoter". To test our model, we used available genome-wide datasets of H3K4 methyltransferases knockouts. Our analysis suggests that CXXC proteins, as readers of non-methylated CpGs would regulate the "seesaw" mechanism that focuses H3K4me3 to unmethylated sites, while being repulsed from H3K4me1 decorated enhancers and CpG island shores.
Conclusions: Our results show that DNA methylation discriminates promoters from enhancers through H3K4me1-H3K4me3 seesaw mechanism, and suggest its possible function in the inheritance of chromatin marks after cell division. Our analyses suggest aberrant formation of promoter-like regions and ectopic transcription of hypomethylated regions of DNA. Such mechanism process can have important implications in biological process in where it has been reported abnormal DNA methylation status such as cancer and aging.
Keywords: Computational epigenomics; DNA methylation; Enhancers; H3K4me1; H3K4me3; Histone modifications; Next generation sequencing; Promoters.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers.Epigenetics Chromatin. 2016 Sep 9;9(1):37. doi: 10.1186/s13072-016-0087-z. eCollection 2016. Epigenetics Chromatin. 2016. PMID: 27617035 Free PMC article.
-
Reversible Regulation of Promoter and Enhancer Histone Landscape by DNA Methylation in Mouse Embryonic Stem Cells.Cell Rep. 2016 Sep 27;17(1):289-302. doi: 10.1016/j.celrep.2016.08.083. Cell Rep. 2016. PMID: 27681438 Free PMC article.
-
The DNA methylation landscape of enhancers in the guinea pig hippocampus.Epigenomics. 2018 Apr 1;10(4):349-365. doi: 10.2217/epi-2017-0064. Epub 2018 Apr 4. Epigenomics. 2018. PMID: 29616589
-
Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation.Biochim Biophys Acta Gene Regul Mech. 2020 Aug;1863(8):194567. doi: 10.1016/j.bbagrm.2020.194567. Epub 2020 Apr 29. Biochim Biophys Acta Gene Regul Mech. 2020. PMID: 32360393 Free PMC article. Review.
-
Studying the epigenome using next generation sequencing.J Med Genet. 2011 Nov;48(11):721-30. doi: 10.1136/jmedgenet-2011-100242. Epub 2011 Aug 8. J Med Genet. 2011. PMID: 21825079 Review.
Cited by
-
Role of Non-Coding Regulatory Elements in the Control of GR-Dependent Gene Expression.Int J Mol Sci. 2021 Apr 20;22(8):4258. doi: 10.3390/ijms22084258. Int J Mol Sci. 2021. PMID: 33923915 Free PMC article.
-
Histone 3 lysine 4 monomethylation supports activation of transcription in S. cerevisiae during nutrient stress.Curr Genet. 2022 Apr;68(2):181-194. doi: 10.1007/s00294-022-01226-2. Epub 2022 Jan 18. Curr Genet. 2022. PMID: 35041077 Free PMC article.
-
A longitudinal and transancestral analysis of DNA methylation patterns and disease activity in lupus patients.JCI Insight. 2020 Nov 19;5(22):e143654. doi: 10.1172/jci.insight.143654. JCI Insight. 2020. PMID: 33108347 Free PMC article.
-
Hypoxia-induced lncHILAR promotes renal cancer metastasis via ceRNA for the miR-613/206/ 1-1-3p/Jagged-1/Notch/CXCR4 signaling pathway.Mol Ther. 2021 Oct 6;29(10):2979-2994. doi: 10.1016/j.ymthe.2021.05.020. Epub 2021 May 29. Mol Ther. 2021. Retraction in: Mol Ther. 2025 Mar 5;33(3):1303. doi: 10.1016/j.ymthe.2025.01.044. PMID: 34058384 Free PMC article. Retracted.
-
Cotton under heat stress: a comprehensive review of molecular breeding, genomics, and multi-omics strategies.Front Genet. 2025 Mar 18;16:1553406. doi: 10.3389/fgene.2025.1553406. eCollection 2025. Front Genet. 2025. PMID: 40171219 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
