

Restoring synaptic plasticity and memory in mouse models of Alzheimer's disease by PKR inhibition
- PMID: 29233183
- PMCID: PMC5727890
- DOI: 10.1186/s13041-017-0338-3
Restoring synaptic plasticity and memory in mouse models of Alzheimer's disease by PKR inhibition
Abstract
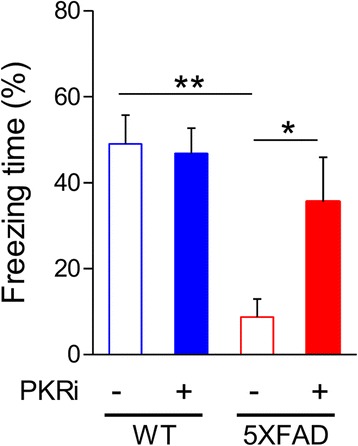
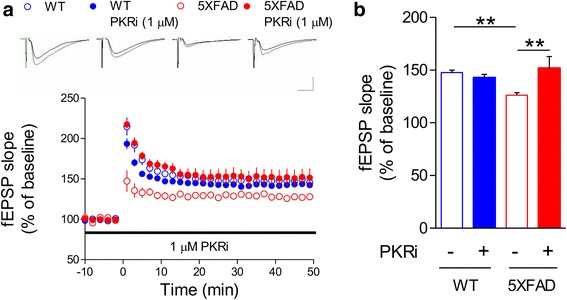
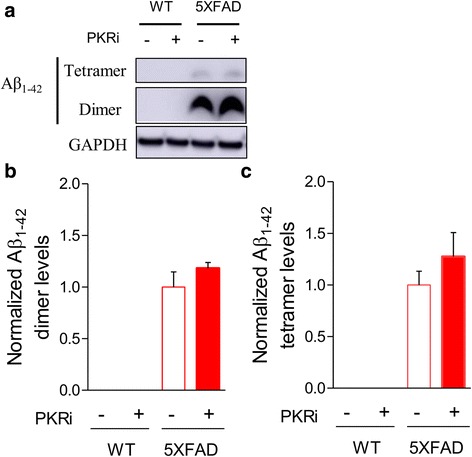
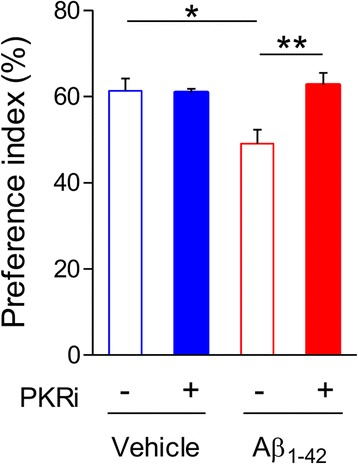
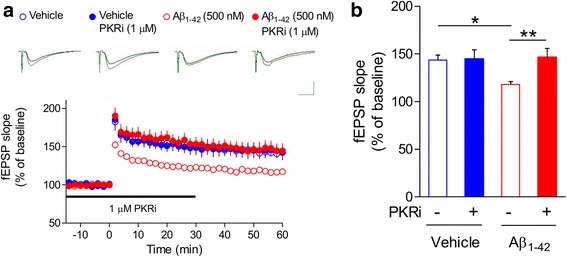
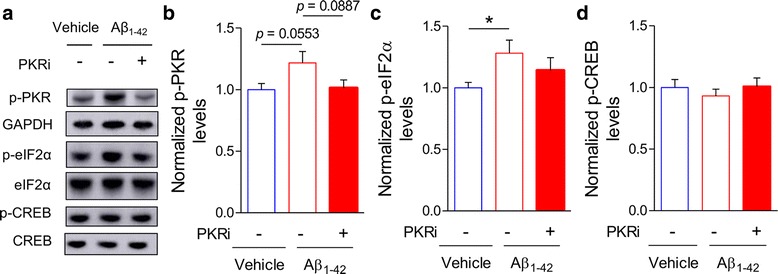
Alzheimer's disease (AD) is a neurodegenerative disorder associated with deficits in cognition and synaptic plasticity. While accumulation of amyloid β (Aβ) and hyper-phosphorylation of tau are parts of the etiology, AD can be caused by a large number of different genetic mutations and other unknown factors. Considering such a heterogeneous nature of AD, it would be desirable to develop treatment strategies that can improve memory irrespective of the individual causes. Reducing the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) was shown to enhance long-term memory and synaptic plasticity in naïve mice. Moreover, hyper-phosphorylation of eIF2α is observed in the brains of postmortem AD patients. Therefore, regulating eIF2α phosphorylation can be a plausible candidate for restoring memory in AD by targeting memory-enhancing mechanism. In this study, we examined whether PKR inhibition can rescue synaptic and learning deficits in two different AD mouse models; 5XFAD transgenic and Aβ1-42-injected mice. We found that the acute treatment of PKR inhibitor (PKRi) can restore the deficits in long-term memory and long-term potentiation (LTP) in both mouse models without affecting the Aβ load in the hippocampus. Our results prove the principle that targeting memory enhancing mechanisms can be a valid candidate for developing AD treatment.
Keywords: Alzheimer’s disease (AD); Amyloid β (Aβ); Contextual fear conditioning; Long-term potentiation (LTP); Object recognition memory; PKR inhibitor (PKRi).
Conflict of interest statement
Ethics approval
All the animal experiments were approved by the Seoul National University Institutional Animal Care and Use Committee (SNU IACUC) and the Chung-Ang University Institutional Animal Care and Use Committee (CAU IACUC).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
