

Dysregulation of autophagy as a common mechanism in lysosomal storage diseases
- PMID: 29233882
- PMCID: PMC5869865
- DOI: 10.1042/EBC20170055
Dysregulation of autophagy as a common mechanism in lysosomal storage diseases
Abstract
The lysosome plays a pivotal role between catabolic and anabolic processes as the nexus for signalling pathways responsive to a variety of factors, such as growth, nutrient availability, energetic status and cellular stressors. Lysosomes are also the terminal degradative organelles for autophagy through which macromolecules and damaged cellular components and organelles are degraded. Autophagy acts as a cellular homeostatic pathway that is essential for organismal physiology. Decline in autophagy during ageing or in many diseases, including late-onset forms of neurodegeneration is considered a major contributing factor to the pathology. Multiple lines of evidence indicate that impairment in autophagy is also a central mechanism underlying several lysosomal storage disorders (LSDs). LSDs are a class of rare, inherited disorders whose histopathological hallmark is the accumulation of undegraded materials in the lysosomes due to abnormal lysosomal function. Inefficient degradative capability of the lysosomes has negative impact on the flux through the autophagic pathway, and therefore dysregulated autophagy in LSDs is emerging as a relevant disease mechanism. Pathology in the LSDs is generally early-onset, severe and life-limiting but current therapies are limited or absent; recognizing common autophagy defects in the LSDs raises new possibilities for therapy. In this review, we describe the mechanisms by which LSDs occur, focusing on perturbations in the autophagy pathway and present the latest data supporting the development of novel therapeutic approaches related to the modulation of autophagy.
Keywords: Autophagy; Glycogenoses; Lysosomal storage disorders; Lysosomes; Neuronal ceroid lipofuscinoses; Sphingolipidoses.
© 2017 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
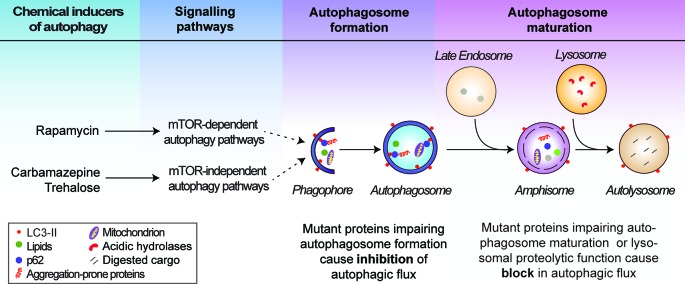
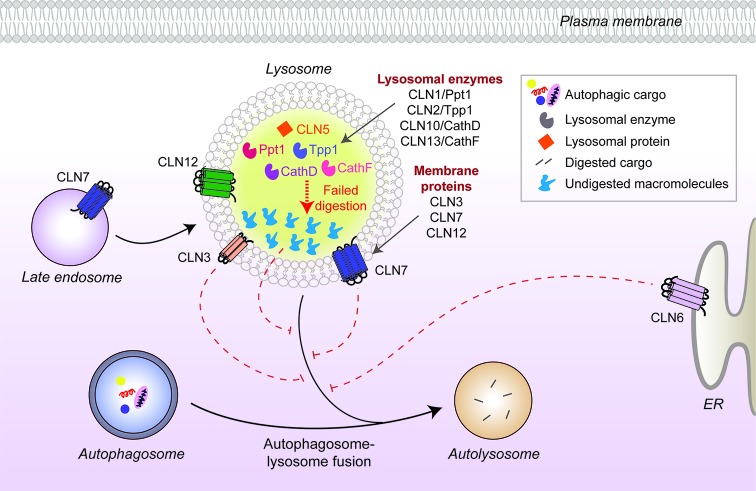
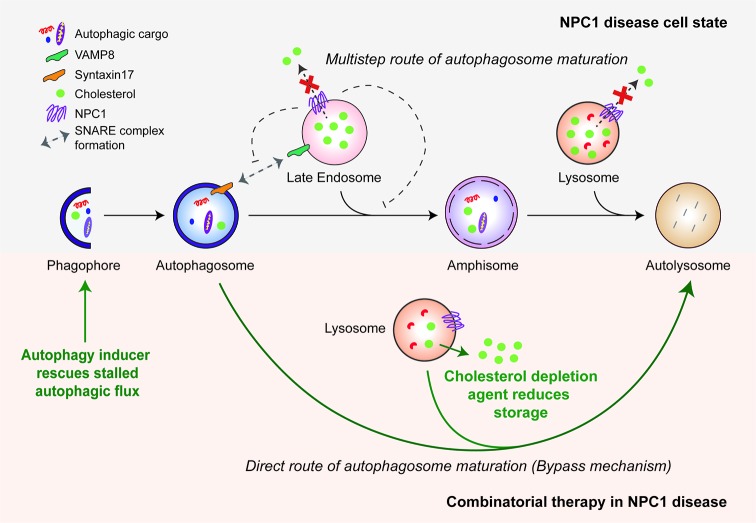
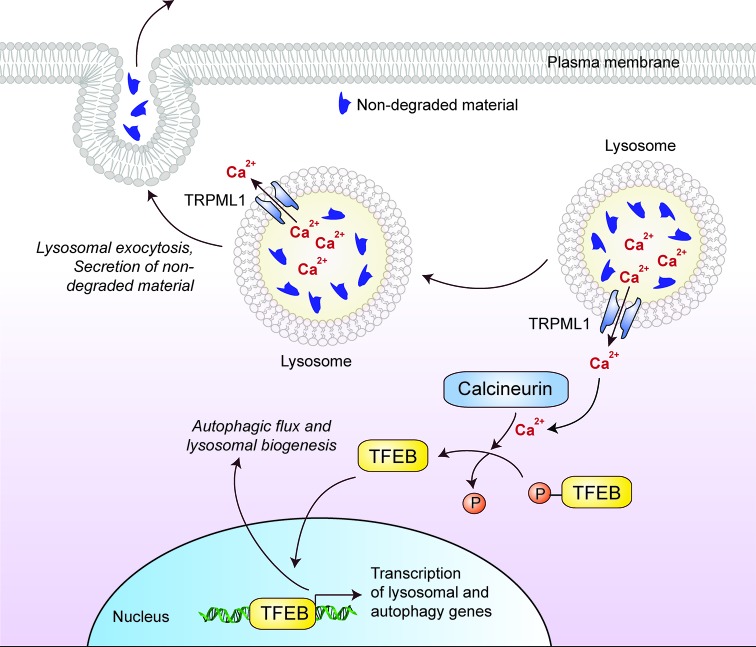
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources