

DNA mismatch repair preferentially protects genes from mutation
- PMID: 29233924
- PMCID: PMC5749183
- DOI: 10.1101/gr.219303.116
DNA mismatch repair preferentially protects genes from mutation
Abstract
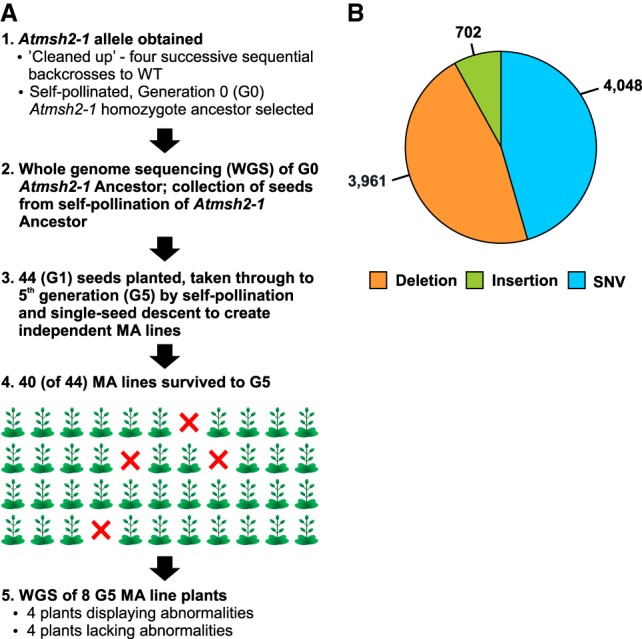
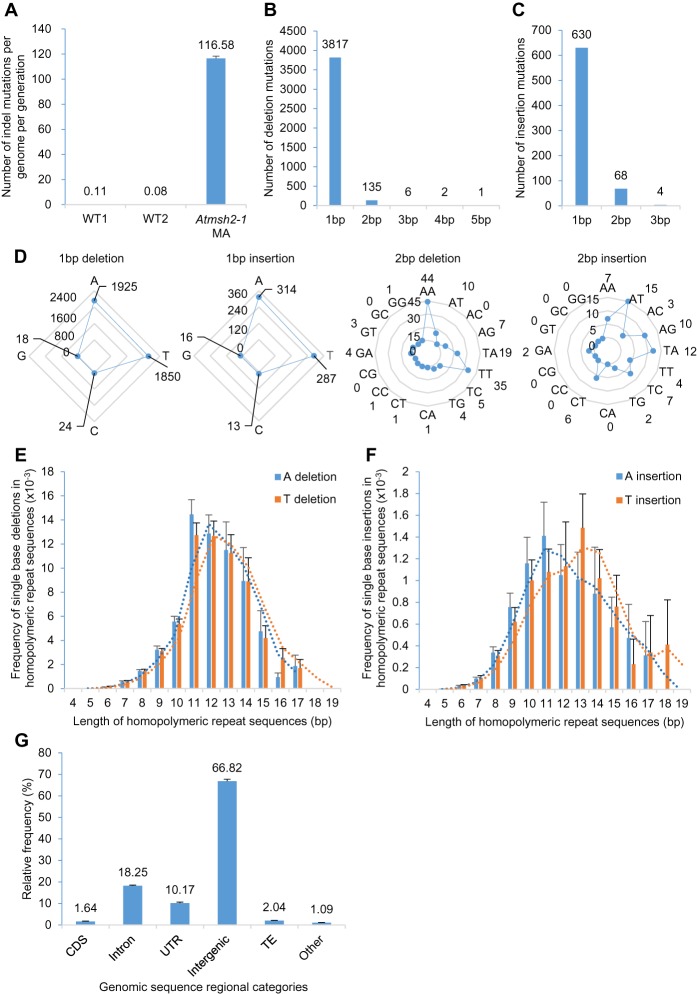
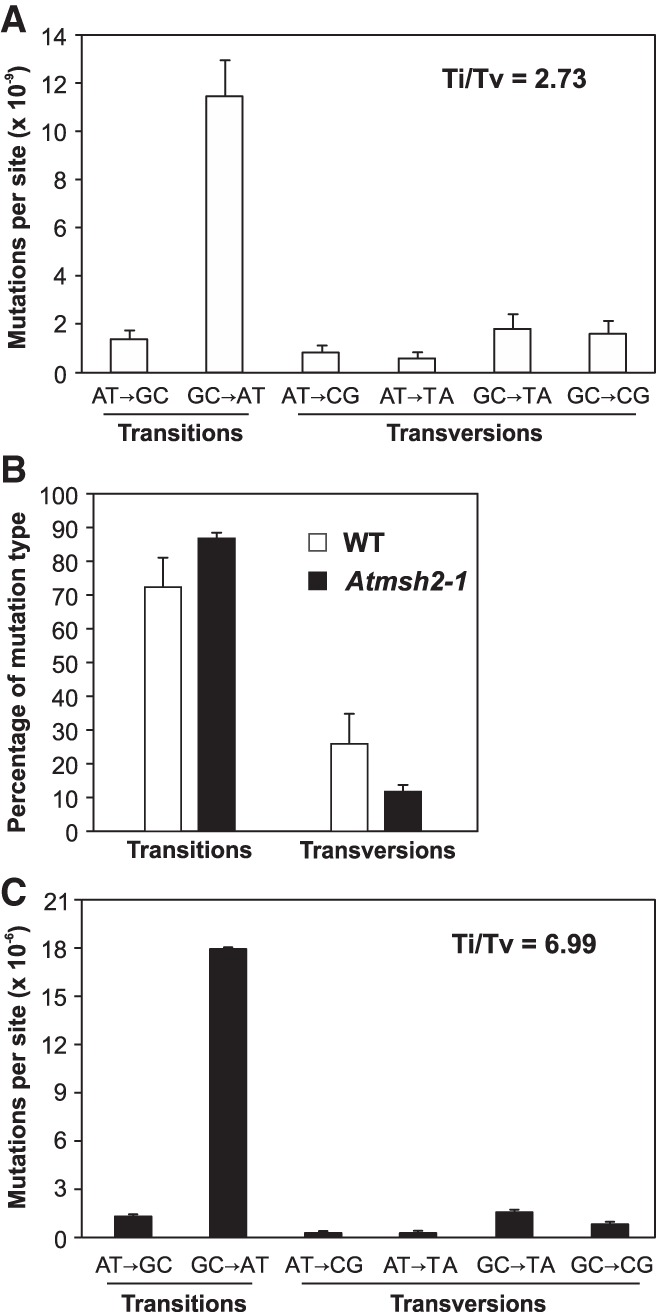
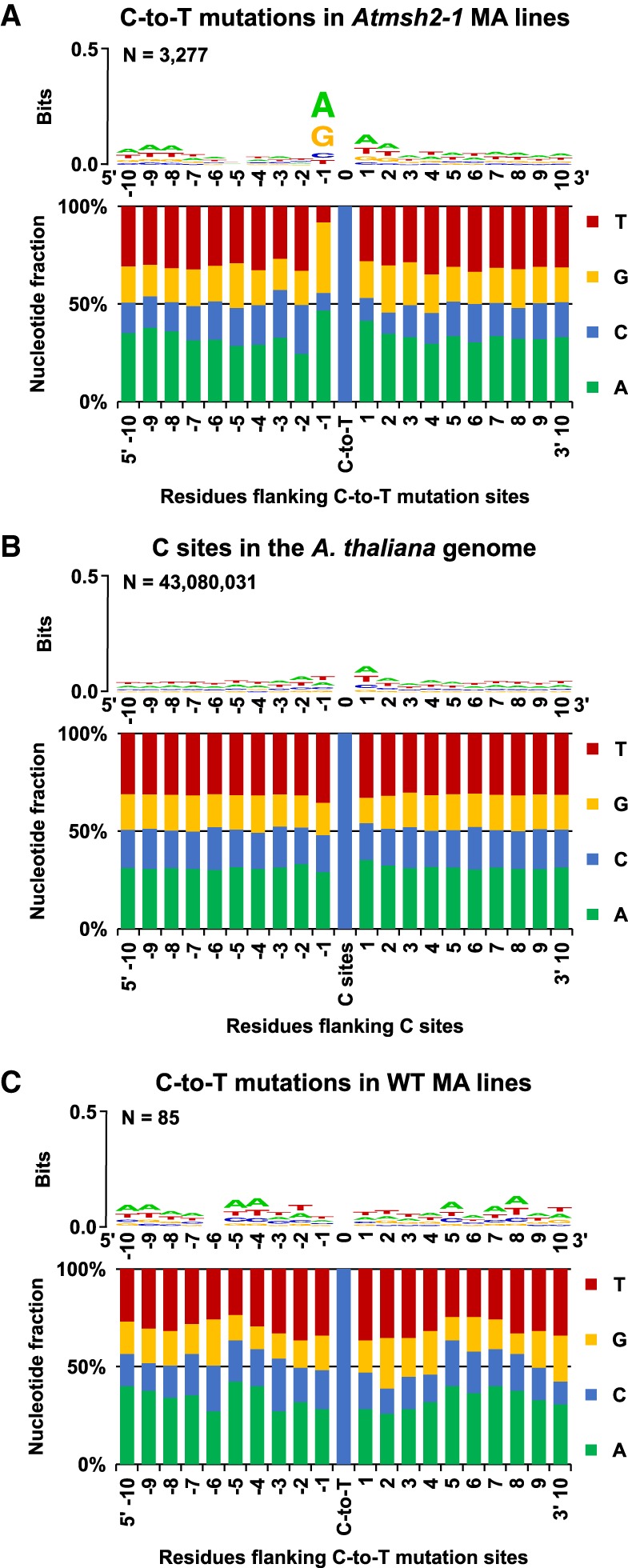
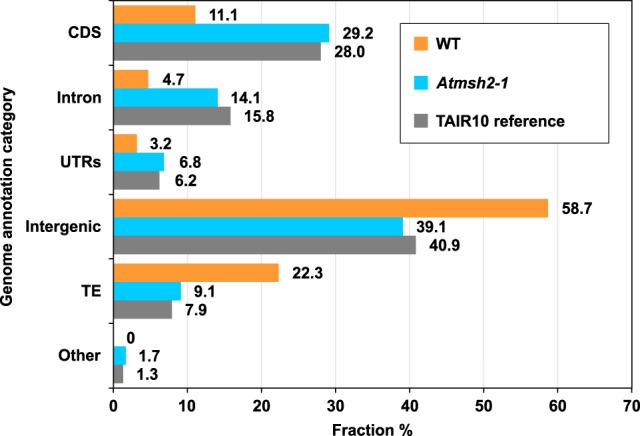
Mutation is the source of genetic variation and fuels biological evolution. Many mutations first arise as DNA replication errors. These errors subsequently evade correction by cellular DNA repair, for example, by the well-known DNA mismatch repair (MMR) mechanism. Here, we determine the genome-wide effects of MMR on mutation. We first identify almost 9000 mutations accumulated over five generations in eight MMR-deficient mutation accumulation (MA) lines of the model plant species, Arabidopsis thaliana We then show that MMR deficiency greatly increases the frequency of both smaller-scale insertions and deletions (indels) and of single-nucleotide variant (SNV) mutations. Most indels involve A or T nucleotides and occur preferentially in homopolymeric (poly A or poly T) genomic stretches. In addition, we find that the likelihood of occurrence of indels in homopolymeric stretches is strongly related to stretch length, and that this relationship causes ultrahigh localized mutation rates in specific homopolymeric stretch regions. For SNVs, we show that MMR deficiency both increases their frequency and changes their molecular mutational spectrum, causing further enhancement of the GC to AT bias characteristic of organisms with normal MMR function. Our final genome-wide analyses show that MMR deficiency disproportionately increases the numbers of SNVs in genes, rather than in nongenic regions of the genome. This latter observation indicates that MMR preferentially protects genes from mutation and has important consequences for understanding the evolution of genomes during both natural selection and human tumor growth.
© 2018 Belfield et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous