

Global unleashing of transcription elongation waves in response to genotoxic stress restricts somatic mutation rate
- PMID: 29233992
- PMCID: PMC5727188
- DOI: 10.1038/s41467-017-02145-4
Global unleashing of transcription elongation waves in response to genotoxic stress restricts somatic mutation rate
Abstract
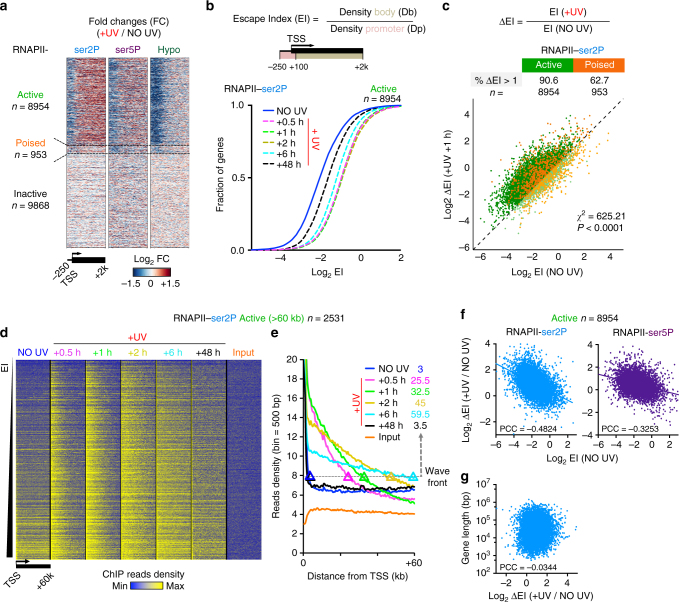
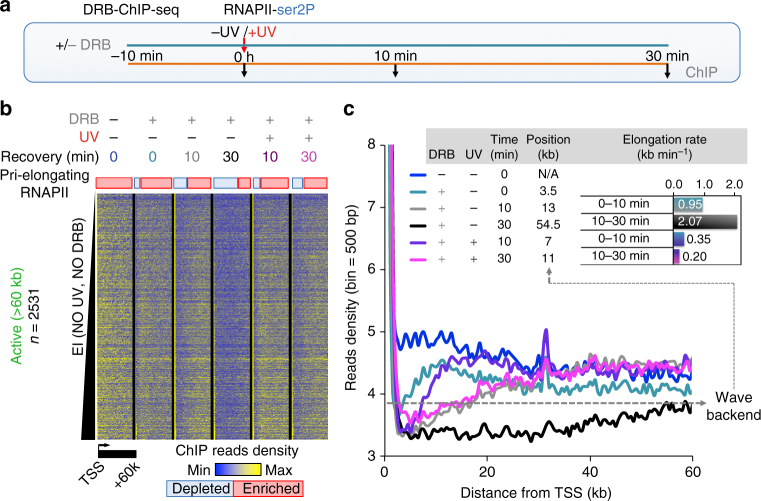
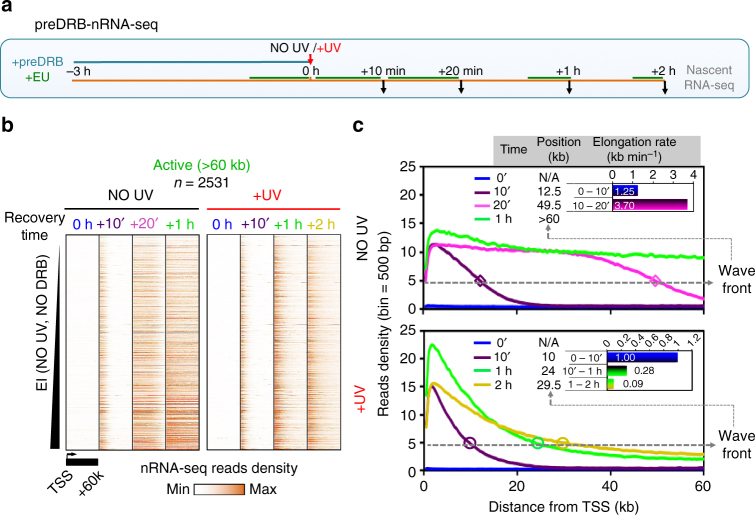
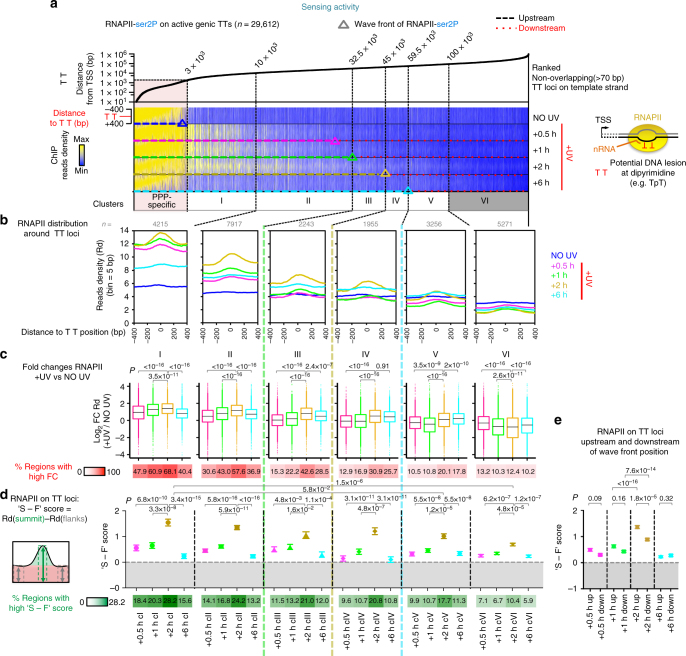
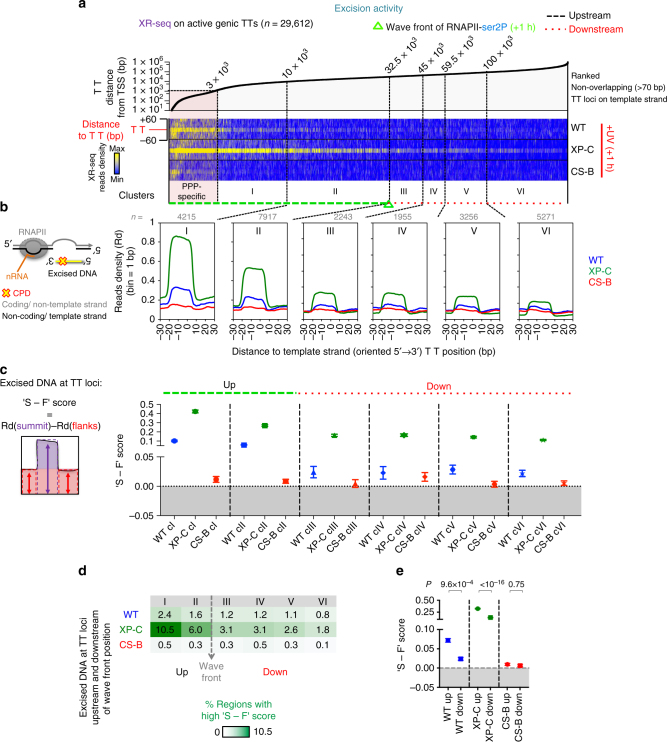
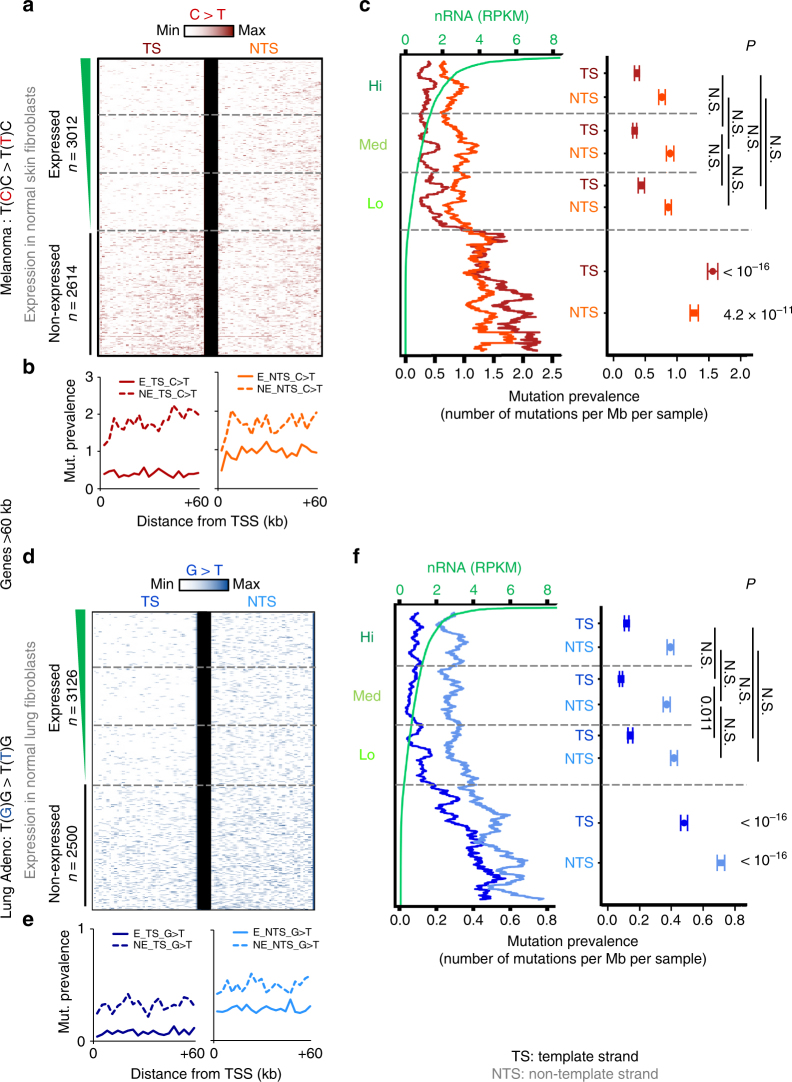
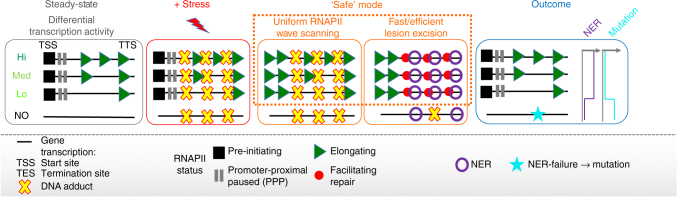
Complex molecular responses preserve gene expression accuracy and genome integrity in the face of environmental perturbations. Here we report that, in response to UV irradiation, RNA polymerase II (RNAPII) molecules are dynamically and synchronously released from promoter-proximal regions into elongation to promote uniform and accelerated surveillance of the whole transcribed genome. The maximised influx of de novo released RNAPII correlates with increased damage-sensing, as confirmed by RNAPII progressive accumulation at dipyrimidine sites and by the average slow-down of elongation rates in gene bodies. In turn, this transcription elongation 'safe' mode guarantees efficient DNA repair regardless of damage location, gene size and transcription level. Accordingly, we detect low and homogenous rates of mutational signatures associated with UV exposure or cigarette smoke across all active genes. Our study reveals a novel advantage for transcription regulation at the promoter-proximal level and provides unanticipated insights into how active transcription shapes the mutagenic landscape of cancer genomes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
