

In situ functional dissection of RNA cis-regulatory elements by multiplex CRISPR-Cas9 genome engineering
- PMID: 29235467
- PMCID: PMC5727397
- DOI: 10.1038/s41467-017-00686-2
In situ functional dissection of RNA cis-regulatory elements by multiplex CRISPR-Cas9 genome engineering
Abstract
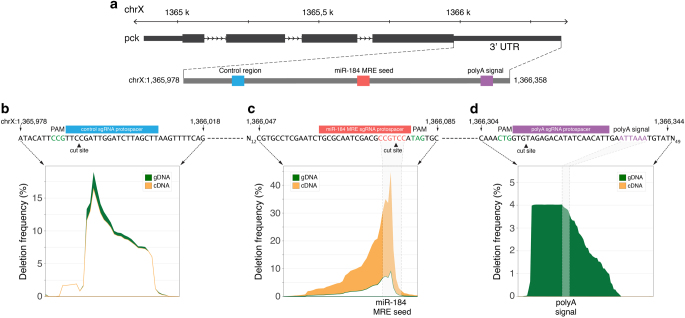
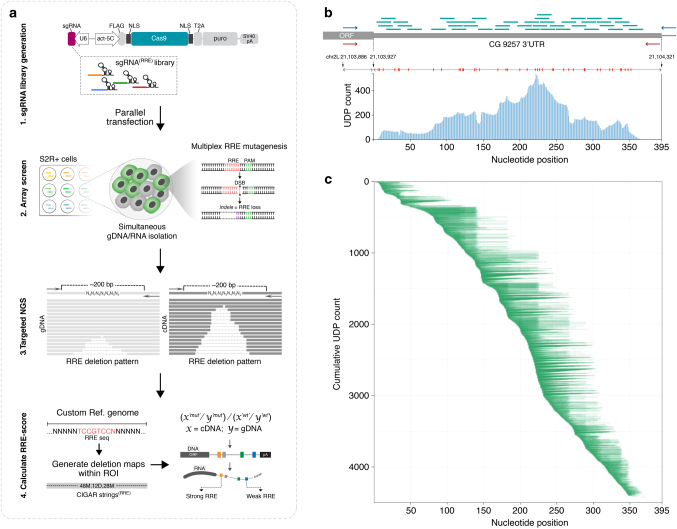
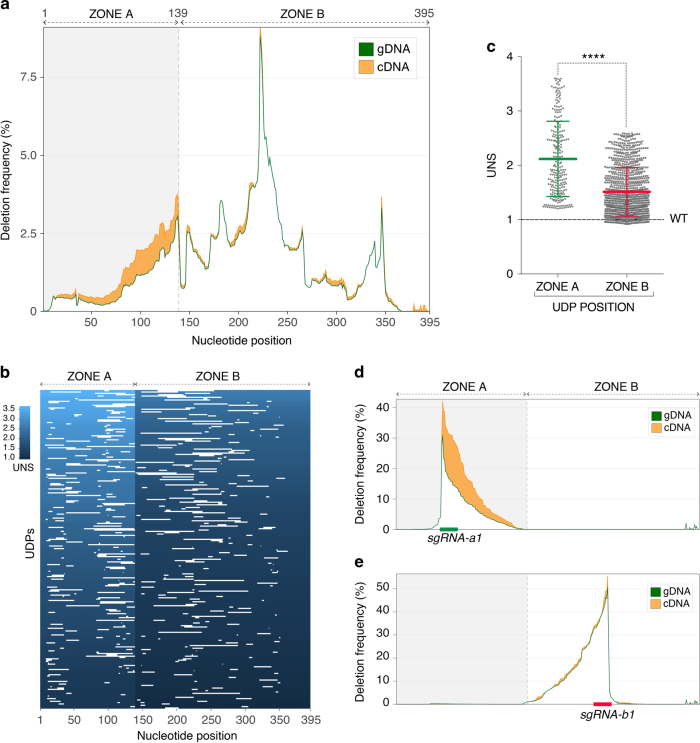
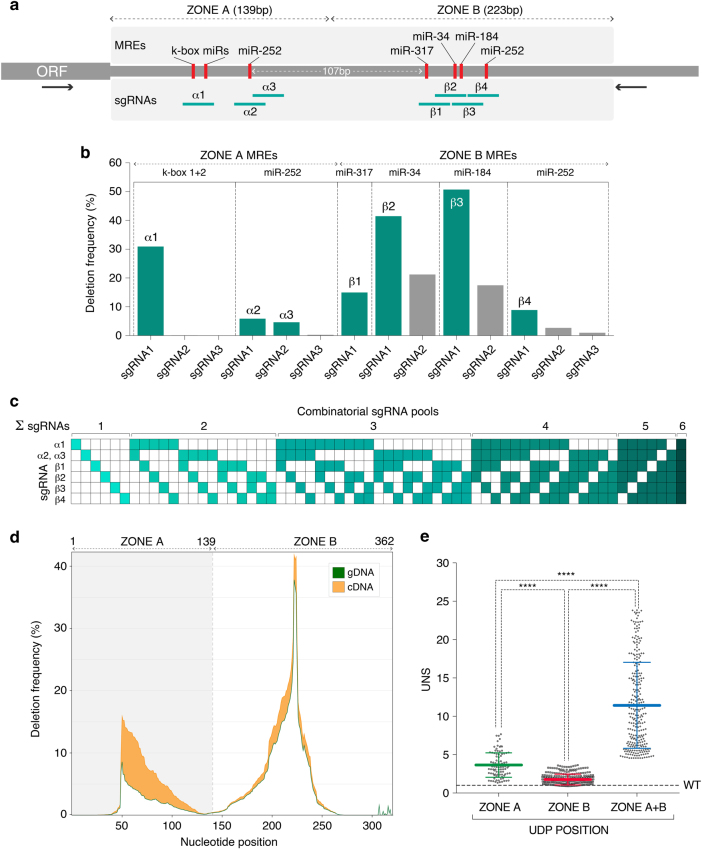
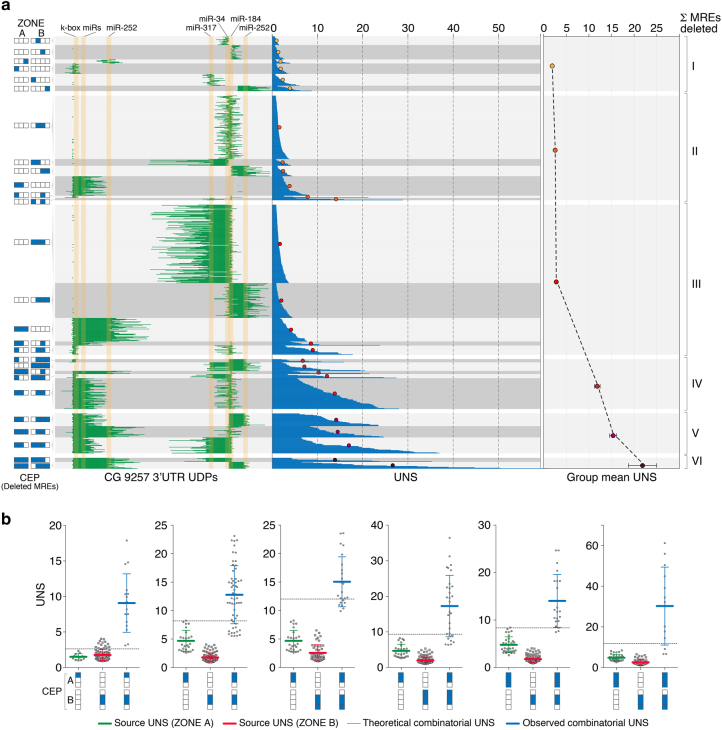
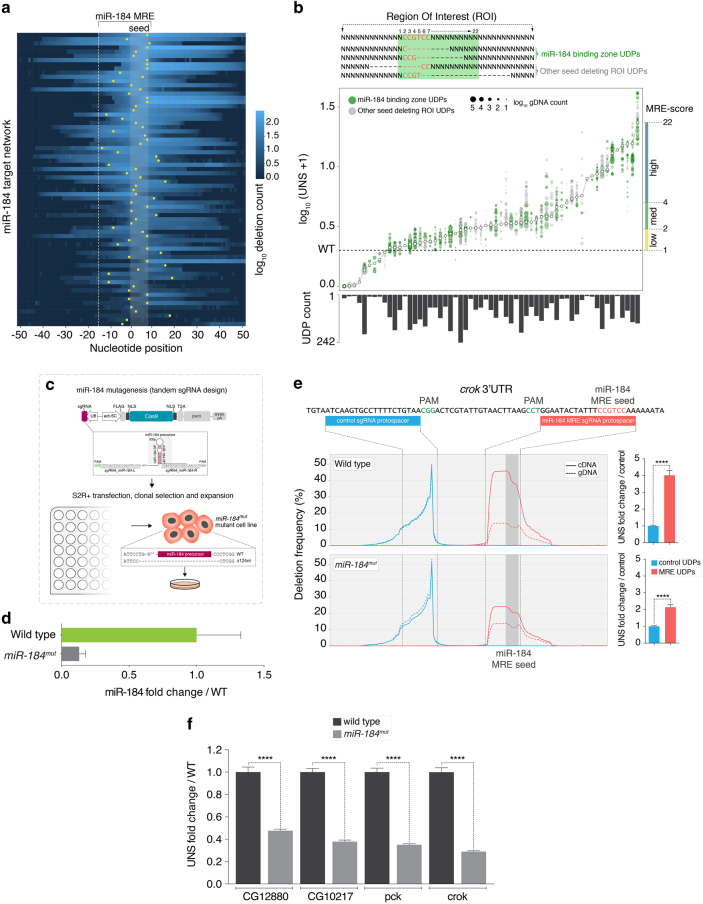
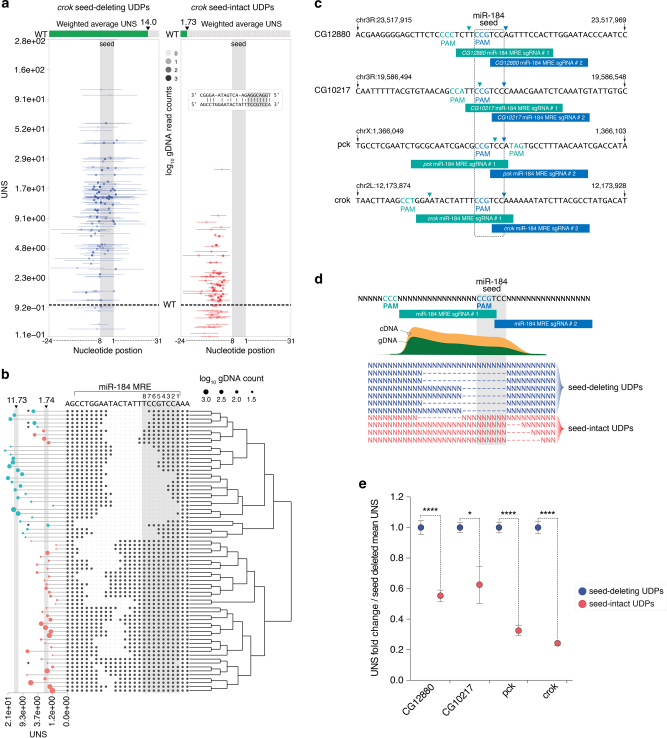
RNA regulatory elements (RREs) are an important yet relatively under-explored facet of gene regulation. Deciphering the prevalence and functional impact of this post-transcriptional control layer requires technologies for disrupting RREs without perturbing cellular homeostasis. Here we describe genome-engineering based evaluation of RNA regulatory element activity (GenERA), a clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 platform for in situ high-content functional analysis of RREs. We use GenERA to survey the entire regulatory landscape of a 3'UTR, and apply it in a multiplex fashion to analyse combinatorial interactions between sets of miRNA response elements (MREs), providing strong evidence for cooperative activity. We also employ this technology to probe the functionality of an entire MRE network under cellular homeostasis, and show that high-resolution analysis of the GenERA dataset can be used to extract functional features of MREs. This study provides a genome editing-based multiplex strategy for direct functional interrogation of RNA cis-regulatory elements in a native cellular environment.
Conflict of interest statement
T.A.M. is one of the founding shareholders of Oxstem Oncology (OSO), a subsidiary company of OxStem Ltd. J.-S.K. is a co-founder and shareholder of ToolGen, Inc., a biotechnology company focused on genome editing. The remaining authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
