

Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40
- PMID: 29236692
- PMCID: PMC5750076
- DOI: 10.1038/nature25023
Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40
Abstract
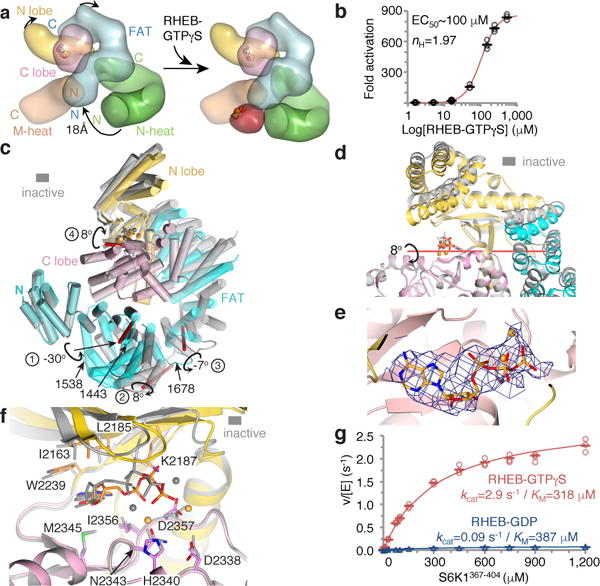
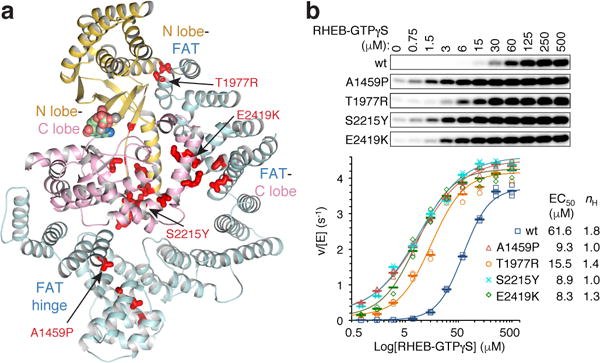
The mechanistic target of rapamycin complex 1 (mTORC1) controls cell growth and metabolism in response to nutrients, energy levels, and growth factors. It contains the atypical kinase mTOR and the RAPTOR subunit that binds to the Tor signalling sequence (TOS) motif of substrates and regulators. mTORC1 is activated by the small GTPase RHEB (Ras homologue enriched in brain) and inhibited by PRAS40. Here we present the 3.0 ångström cryo-electron microscopy structure of mTORC1 and the 3.4 ångström structure of activated RHEB-mTORC1. RHEB binds to mTOR distally from the kinase active site, yet causes a global conformational change that allosterically realigns active-site residues, accelerating catalysis. Cancer-associated hyperactivating mutations map to structural elements that maintain the inactive state, and we provide biochemical evidence that they mimic RHEB relieving auto-inhibition. We also present crystal structures of RAPTOR-TOS motif complexes that define the determinants of TOS recognition, of an mTOR FKBP12-rapamycin-binding (FRB) domain-substrate complex that establishes a second substrate-recruitment mechanism, and of a truncated mTOR-PRAS40 complex that reveals PRAS40 inhibits both substrate-recruitment sites. These findings help explain how mTORC1 selects its substrates, how its kinase activity is controlled, and how it is activated by cancer-associated mutations.
Conflict of interest statement
Figures
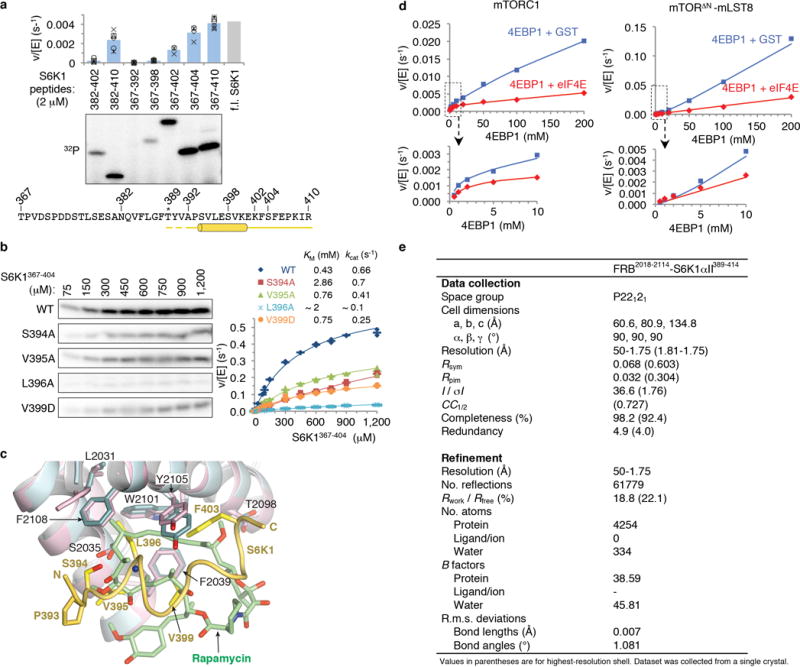
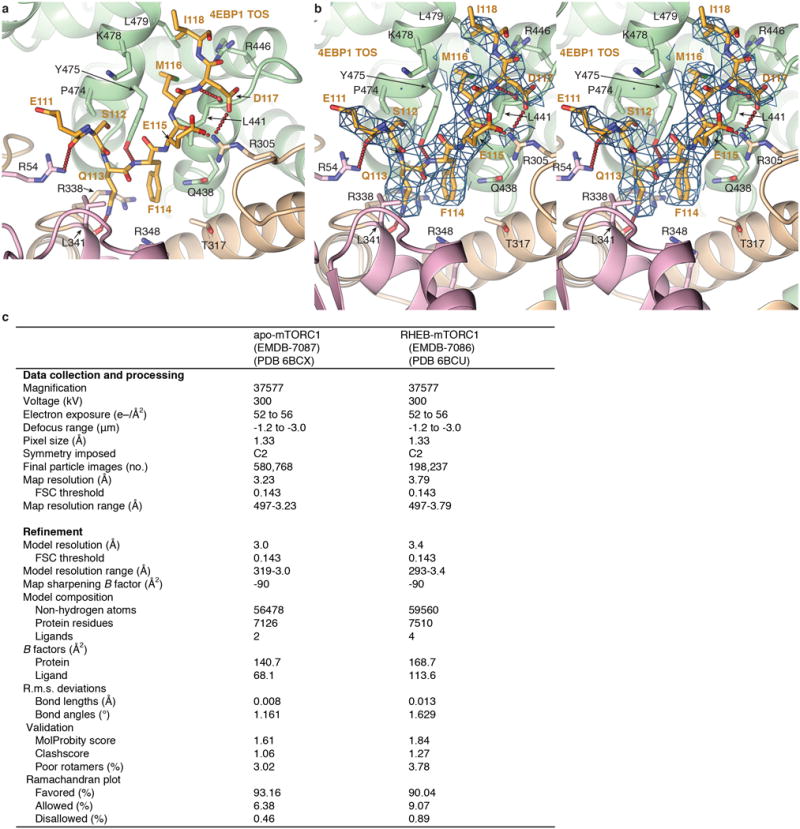
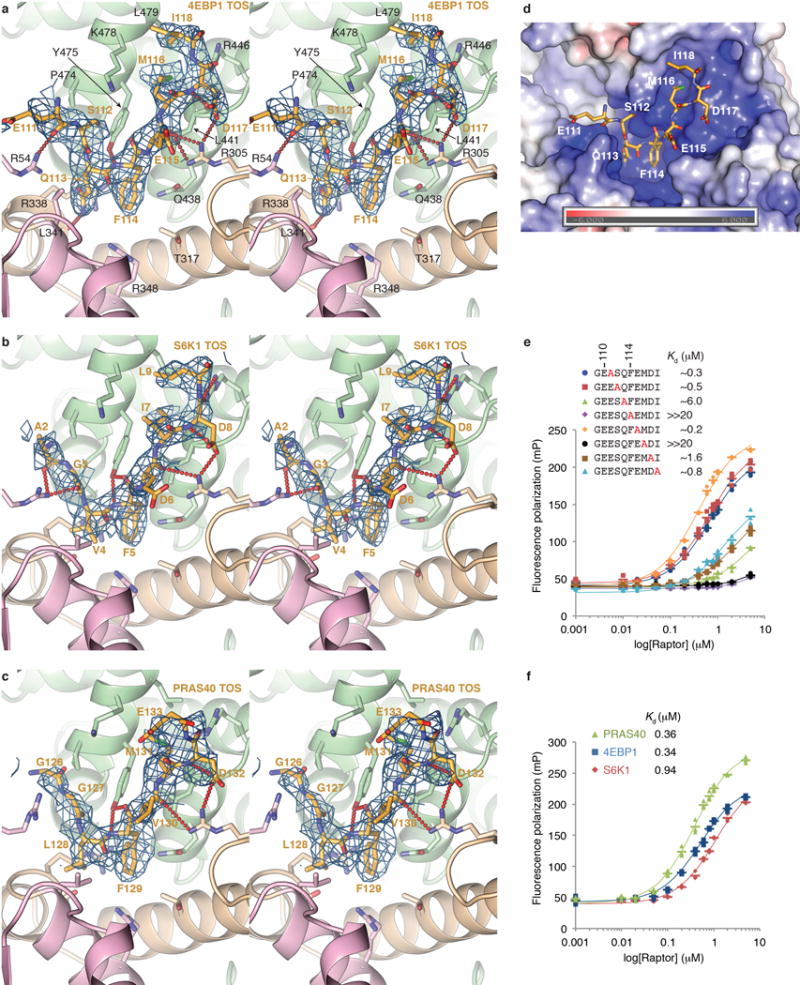
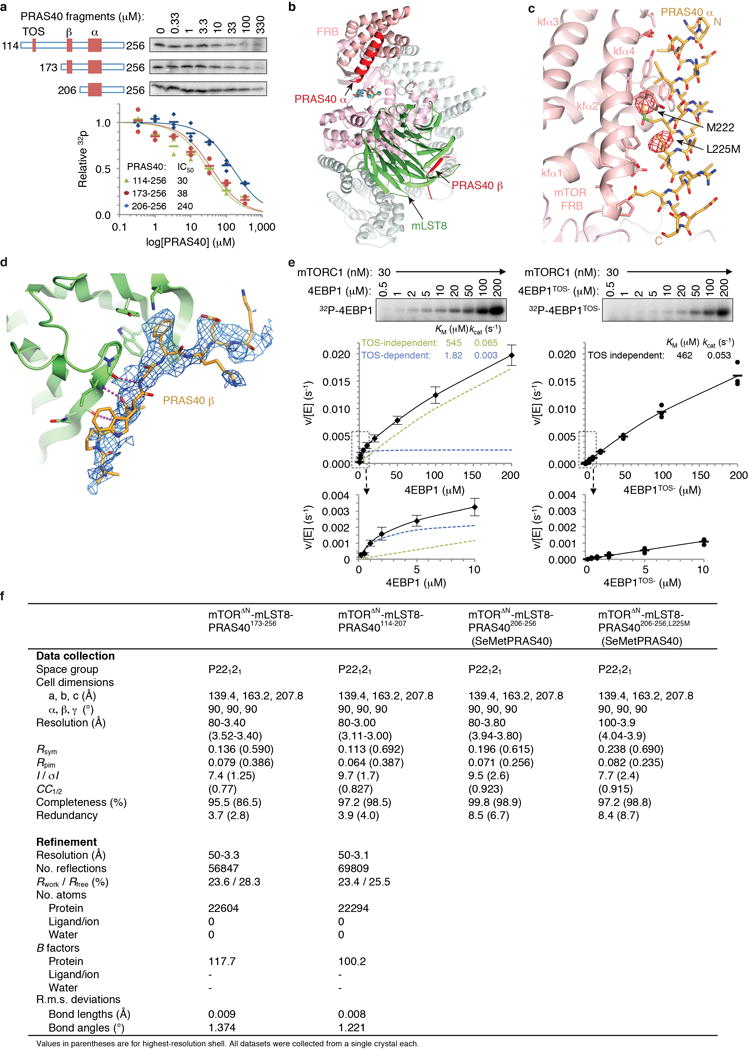
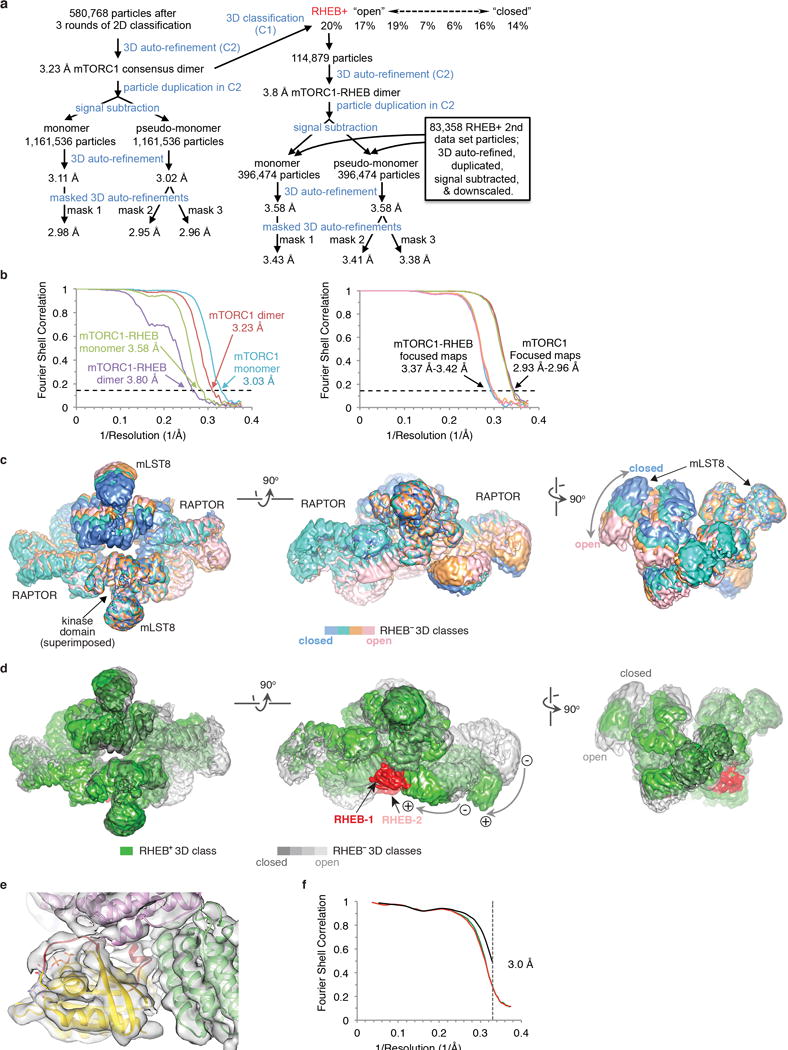

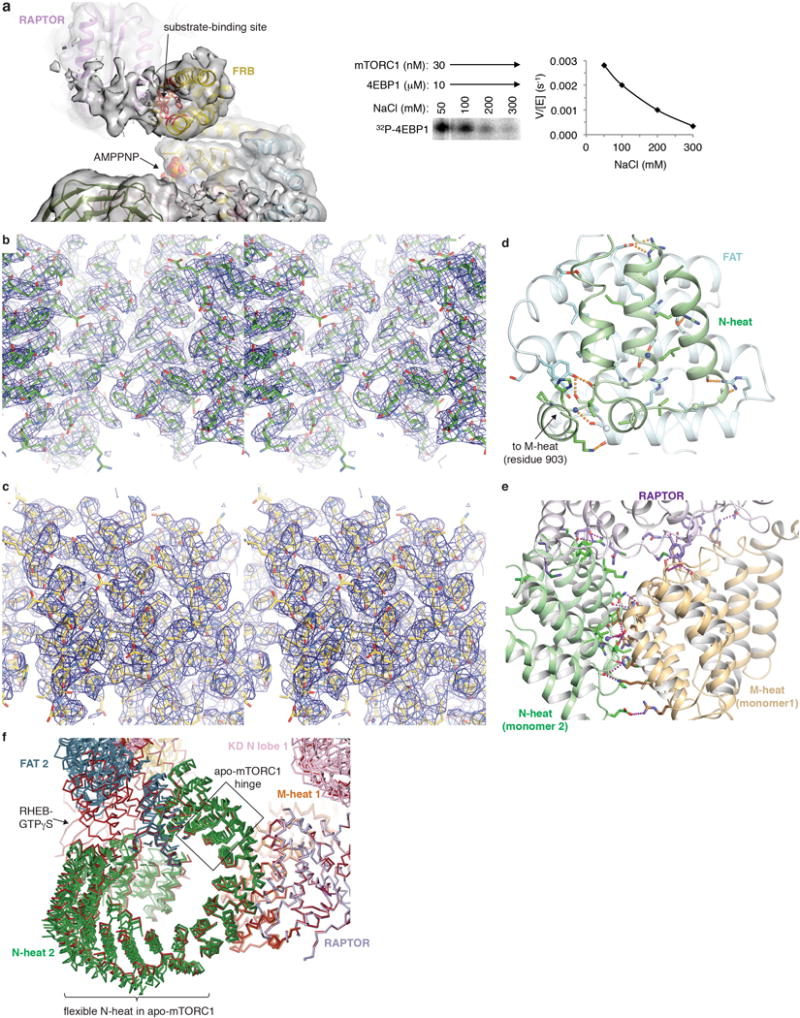
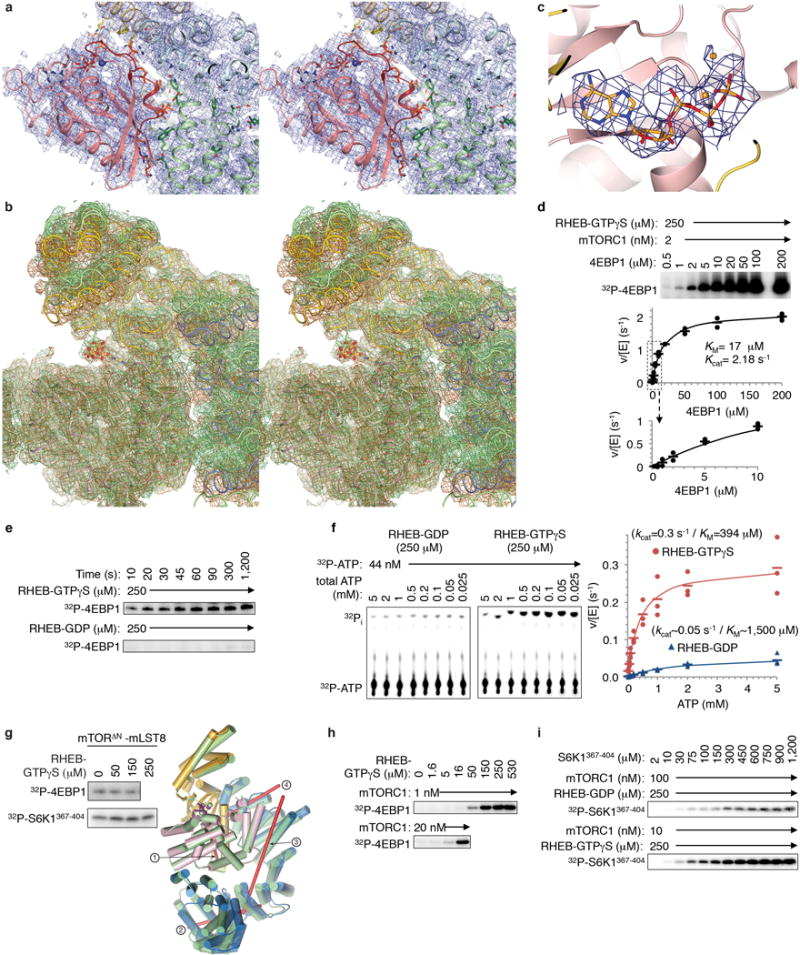
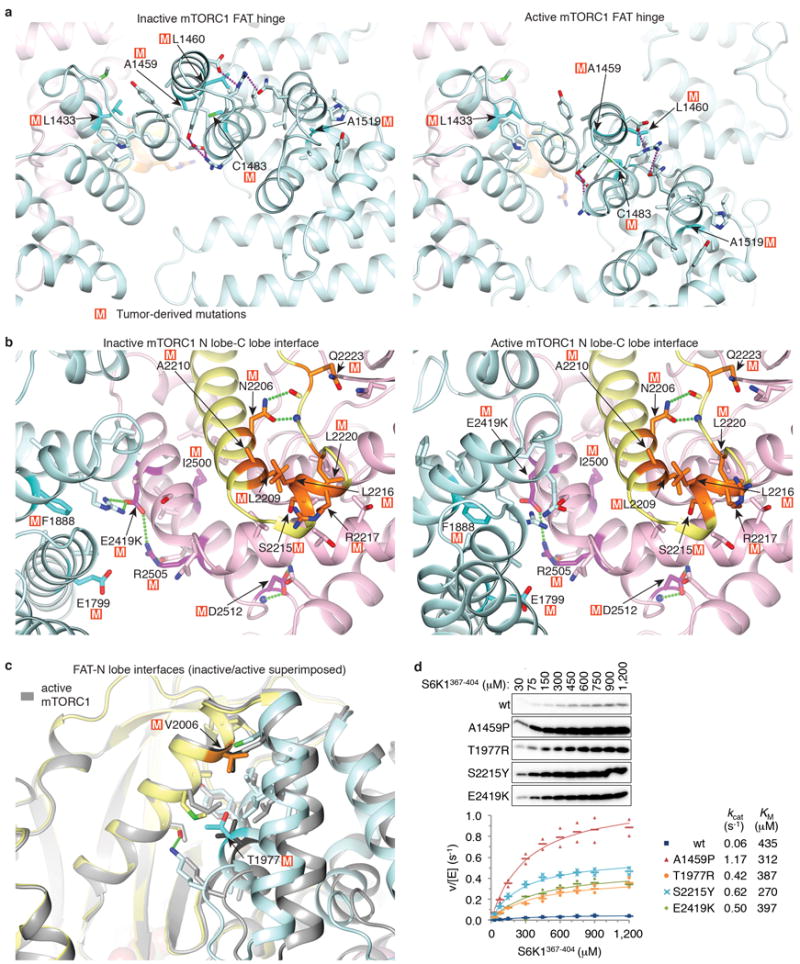
Comment in
-
Determination of mTORC1 Complex Structures Reveals Regulatory Mechanisms.Cancer Discov. 2018 Feb;8(2):OF11. doi: 10.1158/2159-8290.CD-RW2018-001. Epub 2018 Jan 5. Cancer Discov. 2018. PMID: 29305496
References
Extended Data References
-
- Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. - PubMed
-
- Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
