

Potential benefit of bosentan therapy in borderline or less severe pulmonary hypertension secondary to idiopathic pulmonary fibrosis-an interim analysis of results from a prospective, single-center, randomized, parallel-group study
- PMID: 29237441
- PMCID: PMC5729252
- DOI: 10.1186/s12890-017-0523-2
Potential benefit of bosentan therapy in borderline or less severe pulmonary hypertension secondary to idiopathic pulmonary fibrosis-an interim analysis of results from a prospective, single-center, randomized, parallel-group study
Abstract
Background: No drugs have been approved for the treatment of patients with pulmonary hypertension (PH) secondary to idiopathic pulmonary fibrosis (IPF), particularly those with idiopathic honeycomb lung. This study was conducted to investigate the long-term efficacy and safety of bosentan for PH based on changes in prognosis and respiratory failure.
Methods: IPF patients with borderline or less severe PH and completely organized honeycomb lung were randomized (1:1) to bosentan or no treatment for PH for 2 years and assessed at baseline and every 6 months for respiratory failure, activities of daily living (ADL), lung and heart functions by right cardiac catheterization, and other parameters. An interim analysis was performed, however, following detection of a significant survival benefit favoring bosentan therapy.
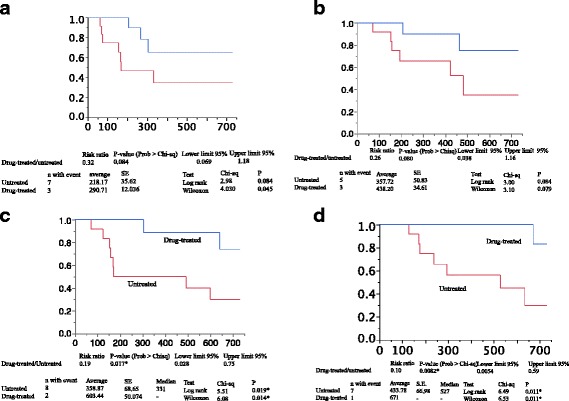
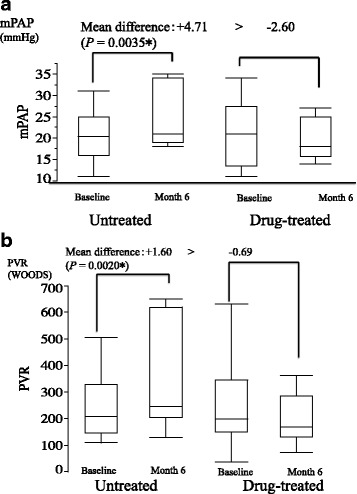
Results: Significant differences were noted for the bosentan-treated (n = 12) vs. untreated (n = 12) groups in hospital-free survival (603.44 ± 50.074 days vs. 358.87 ± 68.65 days; hazard ratio [HR], 0.19; P = 0.017) and overall survival (671 days vs. 433.78 ± 66.98 days; HR, 0.10; P = 0.0082). Again, significant improvements were noted for the bosentan-treated group from baseline to month 6 or 12 in several indices in ADL, pulmonary circulation, and %DLCO. Without requiring O2 inhalation, bosentan was associated with no increase but a trend toward a decrease in adverse events and an improvement in respiratory status.
Conclusions: Bosentan tended to improve prognosis and ADL without worsening respiratory failure in IPF patients with borderline or less severe PH and completely organized honeycomb lung alone.
Trial registration: This study was registered on December 18, 2010 with UMIN-CTR Clinical Trial as UMIN000004749 to investigate the long-term influence of bosentan on cardiac function, as well as its cardioprotective efficacy and safety, in patients with pulmonary hypertension secondary to concurrent COPD and IPF, respectively.
Keywords: Echocardiography; Endothelin receptor antagonists; Idiopathic pulmonary fibrosis; Pulmonary hypertension; Right heart catheterization.
Conflict of interest statement
Ethics approval and consent to participate
An informed consent form describing the following items was prepared. Consent had to be obtained in writing (see Additional file 14; Additional file 15 and Additional file 16). The study protocol was approved by the Ethics Committee of Nippon Medical School. All patients provided their informed consent in writing prior to their participation in this study, and the study was performed in accordance with the ethical standards of the Declaration of Helsinki (2013).
Consent for publication
Not applicable as no personal information was provided in this manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, Mishima M, Kitaichi M, Izumi T. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:650–656. doi: 10.1378/chest.06-1466. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
