

Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin
- PMID: 29237594
- PMCID: PMC5824336
- DOI: 10.1182/blood-2017-05-786590
Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin
Abstract
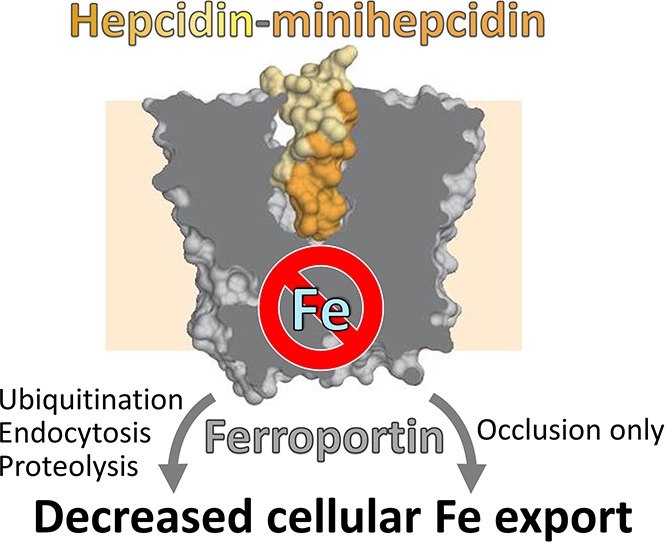
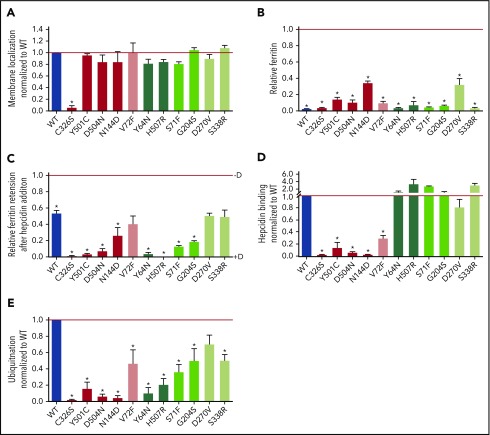
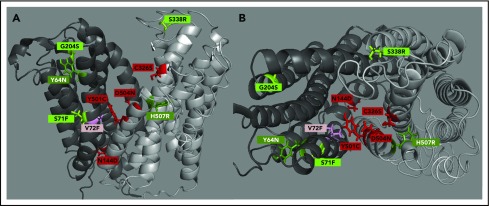
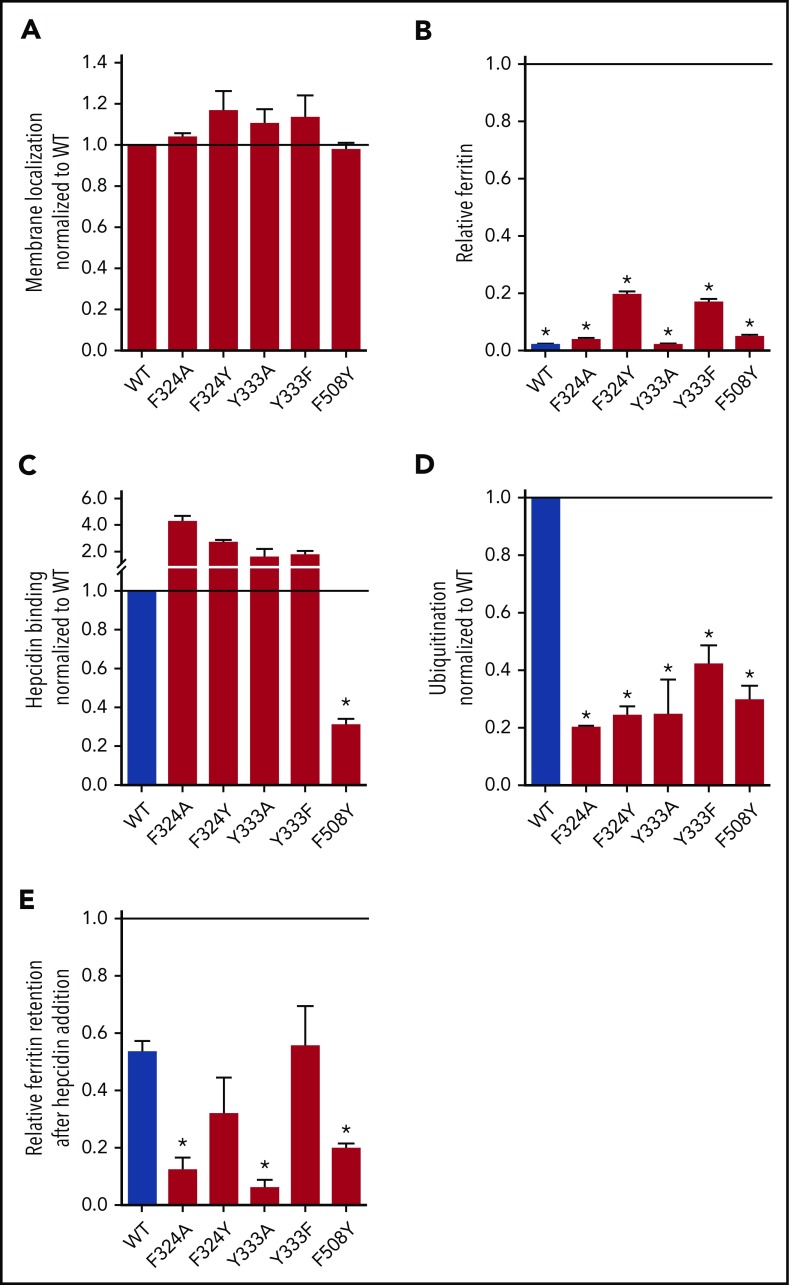
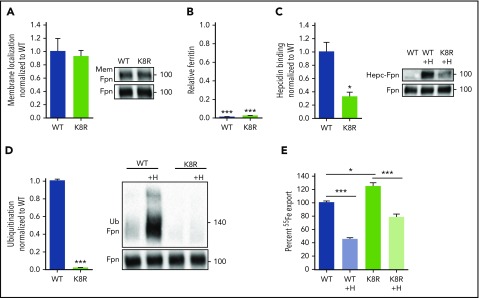
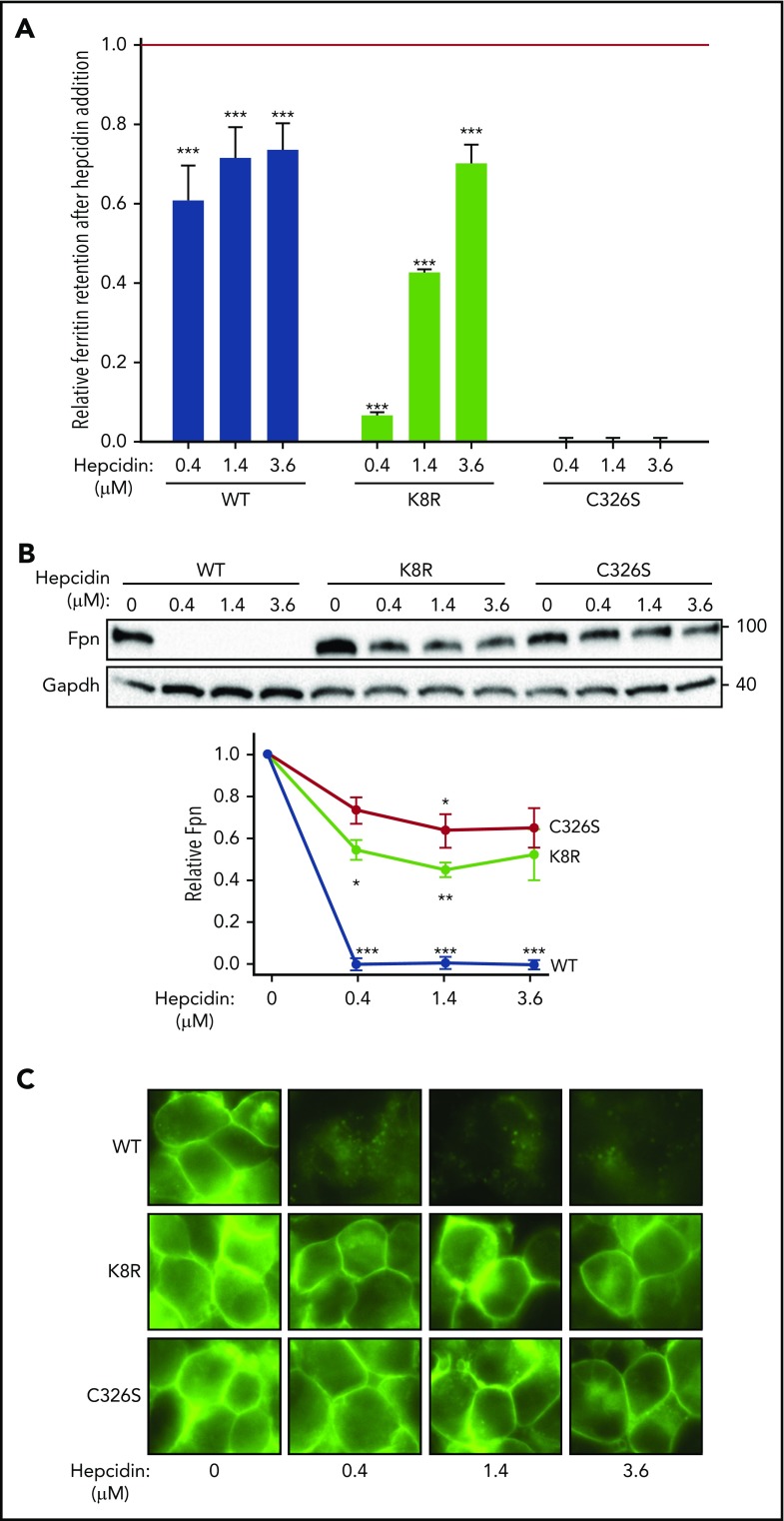
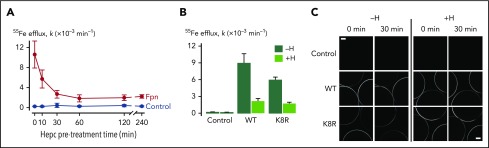
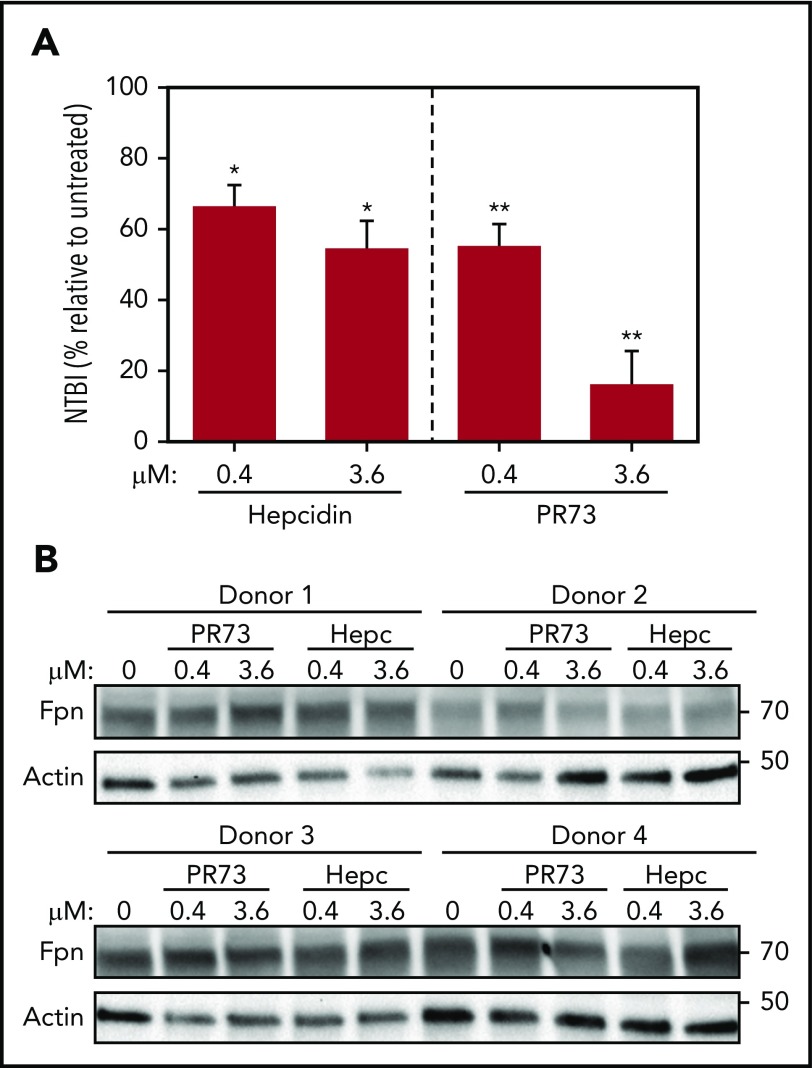
Nonclassical ferroportin disease (FD) is a form of hereditary hemochromatosis caused by mutations in the iron transporter ferroportin (Fpn), resulting in parenchymal iron overload. Fpn is regulated by the hormone hepcidin, which induces Fpn endocytosis and cellular iron retention. We characterized 11 clinically relevant and 5 nonclinical Fpn mutations using stably transfected, inducible isogenic cell lines. All clinical mutants were functionally resistant to hepcidin as a consequence of either impaired hepcidin binding or impaired hepcidin-dependent ubiquitination despite intact hepcidin binding. Mapping the residues onto 2 computational models of the human Fpn structure indicated that (1) mutations that caused ubiquitination-resistance were positioned at helix-helix interfaces, likely preventing the hepcidin-induced conformational change, (2) hepcidin binding occurred within the central cavity of Fpn, (3) hepcidin interacted with up to 4 helices, and (4) hepcidin binding should occlude Fpn and interfere with iron export independently of endocytosis. We experimentally confirmed hepcidin-mediated occlusion of Fpn in the absence of endocytosis in multiple cellular systems: HEK293 cells expressing an endocytosis-defective Fpn mutant (K8R), Xenopus oocytes expressing wild-type or K8R Fpn, and mature human red blood cells. We conclude that nonclassical FD is caused by Fpn mutations that decrease hepcidin binding or hinder conformational changes required for ubiquitination and endocytosis of Fpn. The newly documented ability of hepcidin and its agonists to occlude iron transport may facilitate the development of broadly effective treatments for hereditary iron overload disorders.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: E.N. and T.G. are consultants and shareholders of Intrinsic LifeSciences and Silarus Therapeutics and consultants for La Jolla Pharmaceuticals. T.G. is a consultant for Keryx Pharmaceuticals. B.M. is a recipient of a Vifor Pharma grant. S.R. is a member of the scientific advisory board and the recipient of a grant from Ionis Pharmaceuticals. The remaining authors declare no competing financial interests.
Figures
Comment in
-
How does hepcidin hinder ferroportin activity?Blood. 2018 Feb 22;131(8):840-842. doi: 10.1182/blood-2018-01-824151. Blood. 2018. PMID: 29472371 Free PMC article.
References
-
- Nemeth E, Tuttle MS, Powelson J, et al. . Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
