

Machine learning unifies the modeling of materials and molecules
- PMID: 29242828
- PMCID: PMC5729016
- DOI: 10.1126/sciadv.1701816
Machine learning unifies the modeling of materials and molecules
Abstract
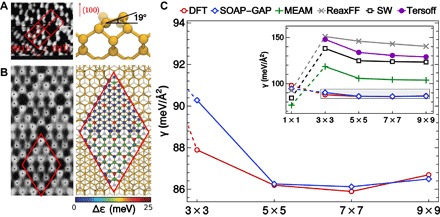
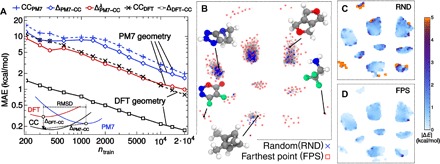
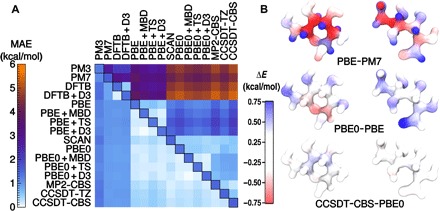
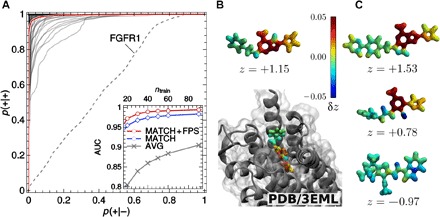
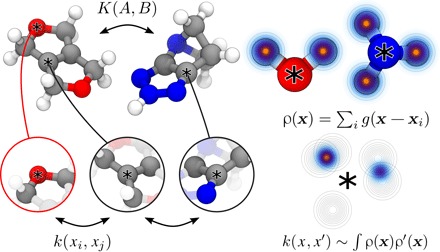
Determining the stability of molecules and condensed phases is the cornerstone of atomistic modeling, underpinning our understanding of chemical and materials properties and transformations. We show that a machine-learning model, based on a local description of chemical environments and Bayesian statistical learning, provides a unified framework to predict atomic-scale properties. It captures the quantum mechanical effects governing the complex surface reconstructions of silicon, predicts the stability of different classes of molecules with chemical accuracy, and distinguishes active and inactive protein ligands with more than 99% reliability. The universality and the systematic nature of our framework provide new insight into the potential energy surface of materials and molecules.
Figures
References
-
- A. Szabo, N. S. Ostlund, Modern Quantum Chemistry (Dover Publications, 2012).
-
- R. M. Martin, Electronic Structure: Basic Theory and Practical Methods (Cambridge Univ. Press, 2004).
-
- Hohenberg P., Kohn W., Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
-
- Kohn W., Sham L. J., Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
-
- Behler J., Parrinello M., Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
