

The ubiquitin-proteasome system is required for African swine fever replication
- PMID: 29244872
- PMCID: PMC5731689
- DOI: 10.1371/journal.pone.0189741
The ubiquitin-proteasome system is required for African swine fever replication
Abstract
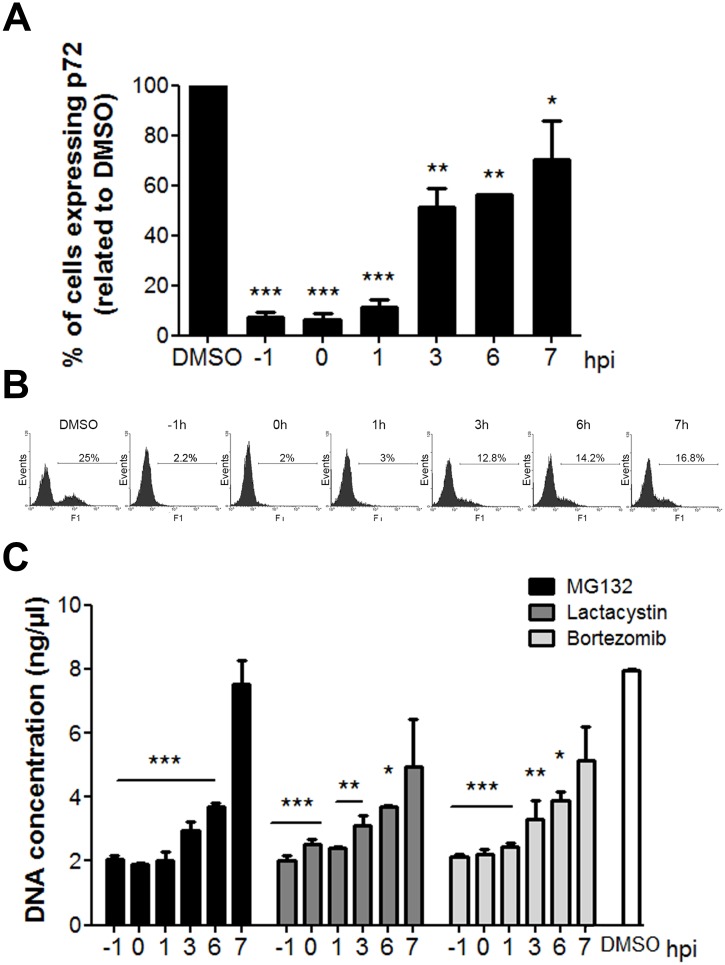
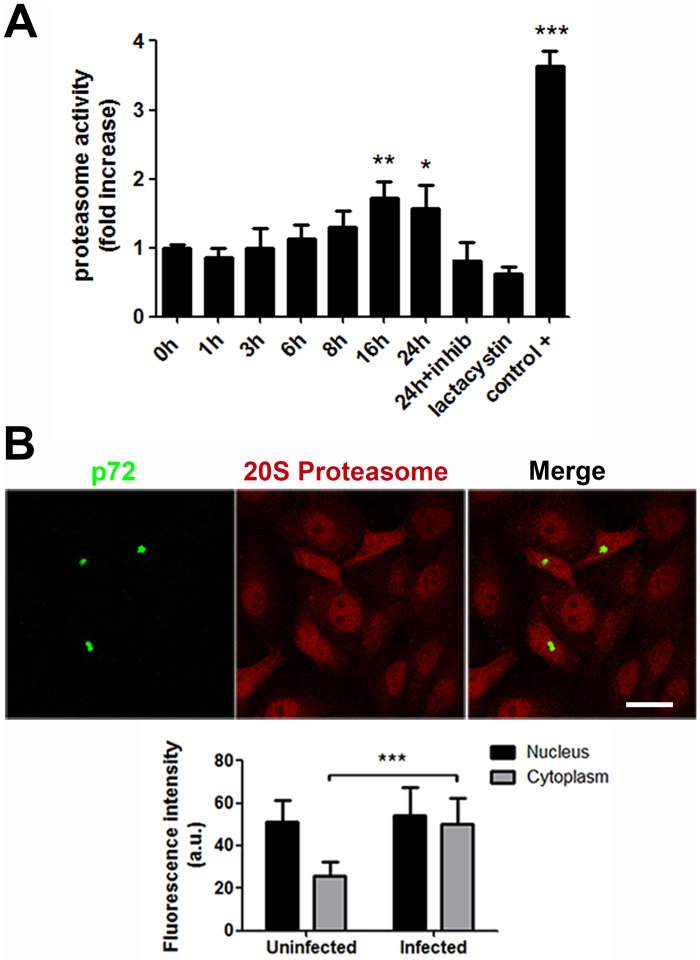
Several viruses manipulate the ubiquitin-proteasome system (UPS) to initiate a productive infection. Determined viral proteins are able to change the host's ubiquitin machinery and some viruses even encode their own ubiquitinating or deubiquitinating enzymes. African swine fever virus (ASFV) encodes a gene homologous to the E2 ubiquitin conjugating (UBC) enzyme. The viral ubiquitin-conjugating enzyme (UBCv1) is expressed throughout ASFV infection and accumulates at late times post infection. UBCv is also present in the viral particle suggesting that the ubiquitin-proteasome pathway could play an important role at early ASFV infection. We determined that inhibition of the final stage of the ubiquitin-proteasome pathway blocked a post-internalization step in ASFV replication in Vero cells. Under proteasome inhibition, ASF viral genome replication, late gene expression and viral production were severely reduced. Also, ASFV enhanced proteasome activity at late times and the accumulation of polyubiquitinated proteins surrounding viral factories. Core-associated and/or viral proteins involved in DNA replication may be targets for the ubiquitin-proteasome pathway that could possibly assist virus uncoating at final core breakdown and viral DNA release. At later steps, polyubiquitinated proteins at viral factories could exert regulatory roles in cell signaling.
Conflict of interest statement
Figures





References
-
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, et al. (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78: 761–771. - PubMed
-
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. Embo J 19: 94–102. doi: 10.1093/emboj/19.1.94 - DOI - PMC - PubMed
-
- Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, et al. (2003) Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem 278: 21323–21326. doi: 10.1074/jbc.C300096200 - DOI - PubMed
-
- Haglund K, Dikic I (2005) Ubiquitylation and cell signaling. Embo J 24: 3353–3359. doi: 10.1038/sj.emboj.7600808 - DOI - PMC - PubMed
-
- Hoeller D, Crosetto N, Blagoev B, Raiborg C, Tikkanen R, et al. (2006) Regulation of ubiquitin-binding proteins by monoubiquitination. Nat Cell Biol 8: 163–169. doi: 10.1038/ncb1354 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
