

Commonalities in epileptogenic processes from different acute brain insults: Do they translate?
- PMID: 29247482
- PMCID: PMC5993212
- DOI: 10.1111/epi.13965
Commonalities in epileptogenic processes from different acute brain insults: Do they translate?
Abstract
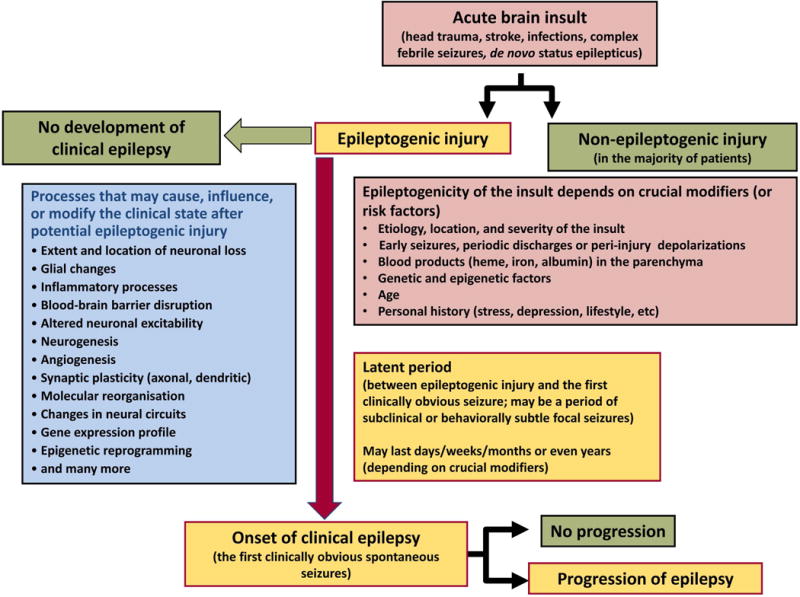
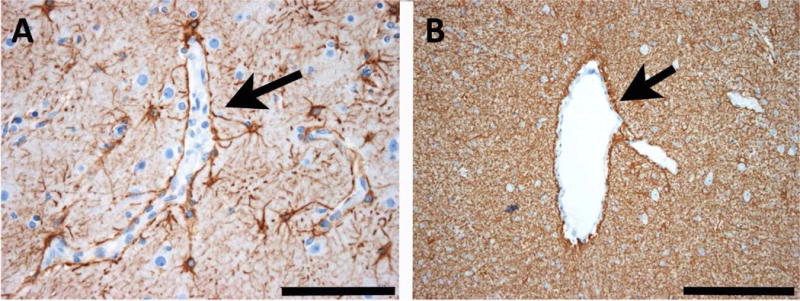
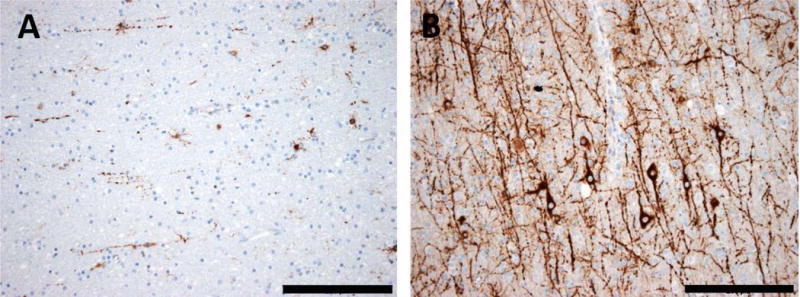
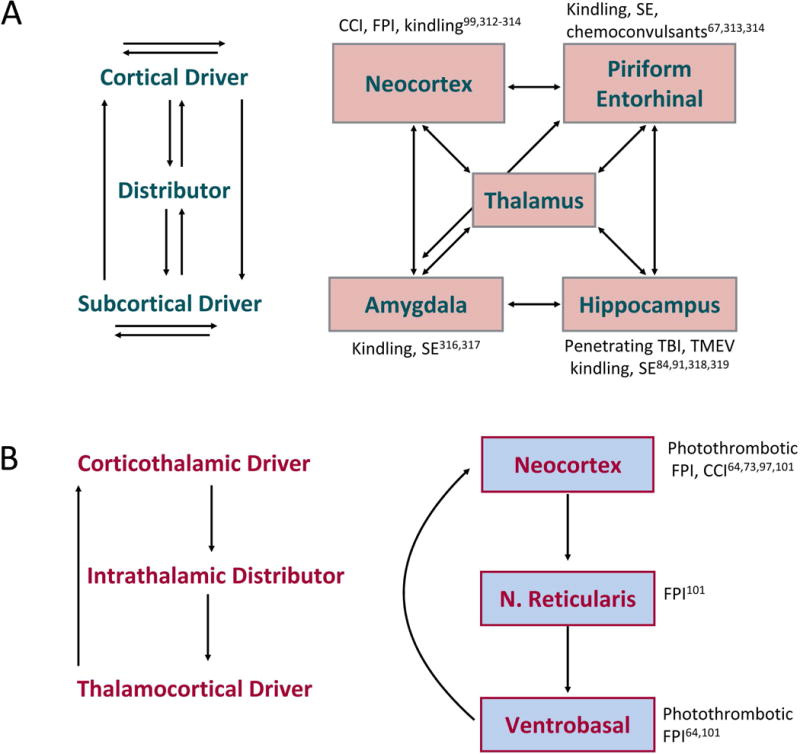
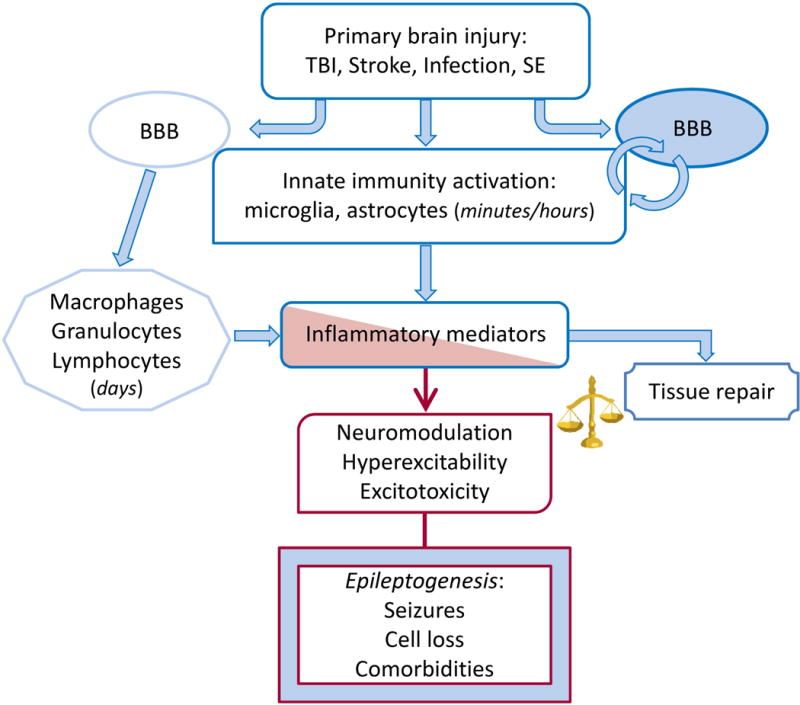
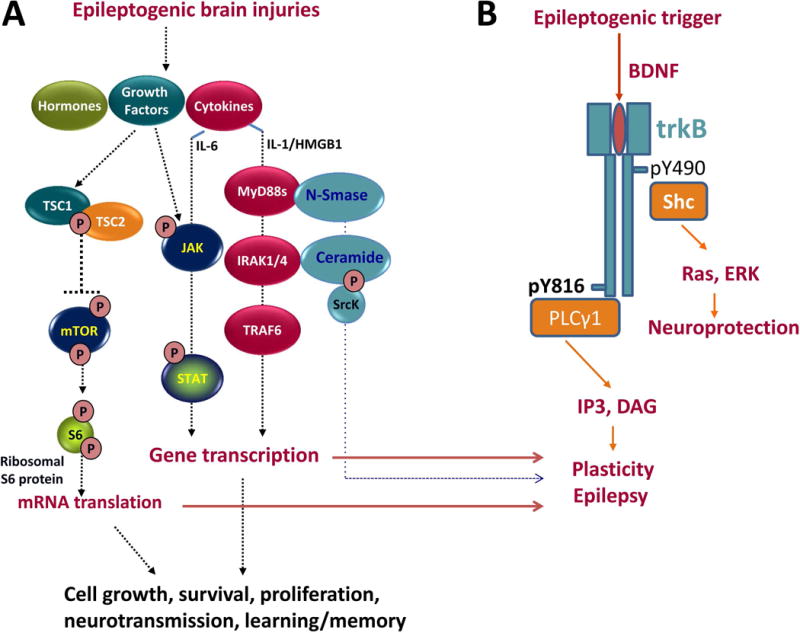
The most common forms of acquired epilepsies arise following acute brain insults such as traumatic brain injury, stroke, or central nervous system infections. Treatment is effective for only 60%-70% of patients and remains symptomatic despite decades of effort to develop epilepsy prevention therapies. Recent preclinical efforts are focused on likely primary drivers of epileptogenesis, namely inflammation, neuron loss, plasticity, and circuit reorganization. This review suggests a path to identify neuronal and molecular targets for clinical testing of specific hypotheses about epileptogenesis and its prevention or modification. Acquired human epilepsies with different etiologies share some features with animal models. We identify these commonalities and discuss their relevance to the development of successful epilepsy prevention or disease modification strategies. Risk factors for developing epilepsy that appear common to multiple acute injury etiologies include intracranial bleeding, disruption of the blood-brain barrier, more severe injury, and early seizures within 1 week of injury. In diverse human epilepsies and animal models, seizures appear to propagate within a limbic or thalamocortical/corticocortical network. Common histopathologic features of epilepsy of diverse and mostly focal origin are microglial activation and astrogliosis, heterotopic neurons in the white matter, loss of neurons, and the presence of inflammatory cellular infiltrates. Astrocytes exhibit smaller K+ conductances and lose gap junction coupling in many animal models as well as in sclerotic hippocampi from temporal lobe epilepsy patients. There is increasing evidence that epilepsy can be prevented or aborted in preclinical animal models of acquired epilepsy by interfering with processes that appear common to multiple acute injury etiologies, for example, in post-status epilepticus models of focal epilepsy by transient treatment with a trkB/PLCγ1 inhibitor, isoflurane, or HMGB1 antibodies and by topical administration of adenosine, in the cortical fluid percussion injury model by focal cooling, and in the albumin posttraumatic epilepsy model by losartan. Preclinical studies further highlight the roles of mTOR1 pathways, JAK-STAT3, IL-1R/TLR4 signaling, and other inflammatory pathways in the genesis or modulation of epilepsy after brain injury. The wealth of commonalities, diversity of molecular targets identified preclinically, and likely multidimensional nature of epileptogenesis argue for a combinatorial strategy in prevention therapy. Going forward, the identification of impending epilepsy biomarkers to allow better patient selection, together with better alignment with multisite preclinical trials in animal models, should guide the clinical testing of new hypotheses for epileptogenesis and its prevention.
Keywords: CNS infections; acquired epilepsy; antiepileptogenesis; epileptogenesis; status epilepticus; stroke; traumatic brain injury.
Wiley Periodicals, Inc. © 2017 International League Against Epilepsy.
Conflict of interest statement
P.K. has served on advisory boards of Lundbeck and UCB Pharma, has acted as a consultant to Eisai, Lundbeck and UCB Pharma, is on Speaker’s Bureau of Eisai, Sunovion and UCB Pharma, and has received research grants from Eisai and Lundbeck. P.L.P. receives royalties from Demos Medical Publishers for the books, Inherited Metabolic Epilepsies and Neuro-Logic: A Primer on Localization. D.B. has acted as consultant to UCB Pharma and Roche. M.J.B. serves on the scientific advisory boards of Eisai, UCB Pharma, GlaxoSmithKline, Lundbeck, Bial, GW Pharmaceuticals and Takeda, is on the speakers’ bureau for Eisai, UCB Pharma, GlaxoSmithKline, Lundbeck, Newbridge, Sanofi Aventis and Abbott and has accepted travel grants for scientific meetings from Eisai, UCB Pharma and Lundbeck. L.J.H. has received research support to Yale University for investigator-initiated studies from Eisai and Upsher-Smith; consultation fees for advising from Ceribell, Monteris, Neuropace, Sun Neuroscience, and Engage Therapeutics; royalties for authoring chapters for UpToDate-Neurology, chapters for Medlink—Neurology, and from Wiley for co-authoring the book “Atlas of EEG in Critical Care”, by Hirsch and Brenner; and honoraria for speaking from Neuropace. A.P. has received research funding from Neurotrauma Sciences LLC. A.V. has acted as consultant to UCB Pharma and Takeda. H.K. and R.M.K. are employees of UCB Pharma. M.C.W. has served on advisory boards of Eisai, and UCB Pharma, has acted as a consultant to GSK, Pfizer, is on Speaker’s Bureau of Eisai, UCB Pharma and SAGE, and has received research grants from Vitaflo. W.L. has served on advisory boards of Grünenthal and UCB Pharma, has acted as a consultant to Pfizer, Lundbeck, GSK, Schering Plough, Boehringer-Ingelheim, UCB Pharma, Bayer, and AWD/Elbion, and has received research grants from UCB Pharma, AWD/Elbion, Merz, Boehringer-Ingelheim, Johnson & Johnson, Pfizer, Novartis, Merz, Desitin, and Piqur. ABK has served on the Executive Committee and Board of American Epilepsy Society and Scientific Advisory Board of CURE, and has acted as a consultant for Pfizer. M.S., J.E.Jr., R.D., I.B., E.A., C.S., R.S.S., C.B., P.A.F., K.K., D.H.L., N.P., M.A.R. and D.S. report no conflict of interest. The mentioned companies or agencies had no role in the design, preparation or writing of this manuscript. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this review is consistent with those guidelines.
Figures

References
-
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1993. Epilepsia. 34:453–68. - PubMed
-
- Löscher W, Klitgaard H, Twyman RE, et al. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–76. - PubMed
-
- Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–3. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 NS103740/NS/NINDS NIH HHS/United States
- R56 NS065877/NS/NINDS NIH HHS/United States
- MR/L01095X/1/MRC_/Medical Research Council/United Kingdom
- U01 NS058158/NS/NINDS NIH HHS/United States
- U54 NS100064/NS/NINDS NIH HHS/United States
- UL1 TR001409/TR/NCATS NIH HHS/United States
- KL2 TR001432/TR/NCATS NIH HHS/United States
- G0802158/MRC_/Medical Research Council/United Kingdom
- R01 NS051710/NS/NINDS NIH HHS/United States
- R37 NS033310/NS/NINDS NIH HHS/United States
- P01 NS002808/NS/NINDS NIH HHS/United States
- R01 NS084920/NS/NINDS NIH HHS/United States
- R21 NS049592/NS/NINDS NIH HHS/United States
- MR/P025641/1/MRC_/Medical Research Council/United Kingdom
- G116/147/MRC_/Medical Research Council/United Kingdom
- R01 NS033310/NS/NINDS NIH HHS/United States
- L40 NS103805/NS/NINDS NIH HHS/United States
- U01 NS042372/NS/NINDS NIH HHS/United States
- R01 NS065957/NS/NINDS NIH HHS/United States
- G0400136/MRC_/Medical Research Council/United Kingdom
- R01 NS061844/NS/NINDS NIH HHS/United States
- U54 HD090257/HD/NICHD NIH HHS/United States
- 083163/WT_/Wellcome Trust/United Kingdom
- R21 NS083057/NS/NINDS NIH HHS/United States
- P20 NS080181/NS/NINDS NIH HHS/United States
- R01 NS082286/NS/NINDS NIH HHS/United States
- U54 NS083932/NS/NINDS NIH HHS/United States
- R21 NS088024/NS/NINDS NIH HHS/United States
- R01 NS097776/NS/NINDS NIH HHS/United States
- RF1 NS033310/NS/NINDS NIH HHS/United States
- R01 NS097762/NS/NINDS NIH HHS/United States
- R01 NS065877/NS/NINDS NIH HHS/United States
- R01 NS071048/NS/NINDS NIH HHS/United States
- 212285/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- U54 HD090255/HD/NICHD NIH HHS/United States
- U54 NS079202/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
