

Pharmacogenomics of GPCR Drug Targets
- PMID: 29249361
- PMCID: PMC5766829
- DOI: 10.1016/j.cell.2017.11.033
Pharmacogenomics of GPCR Drug Targets
Abstract
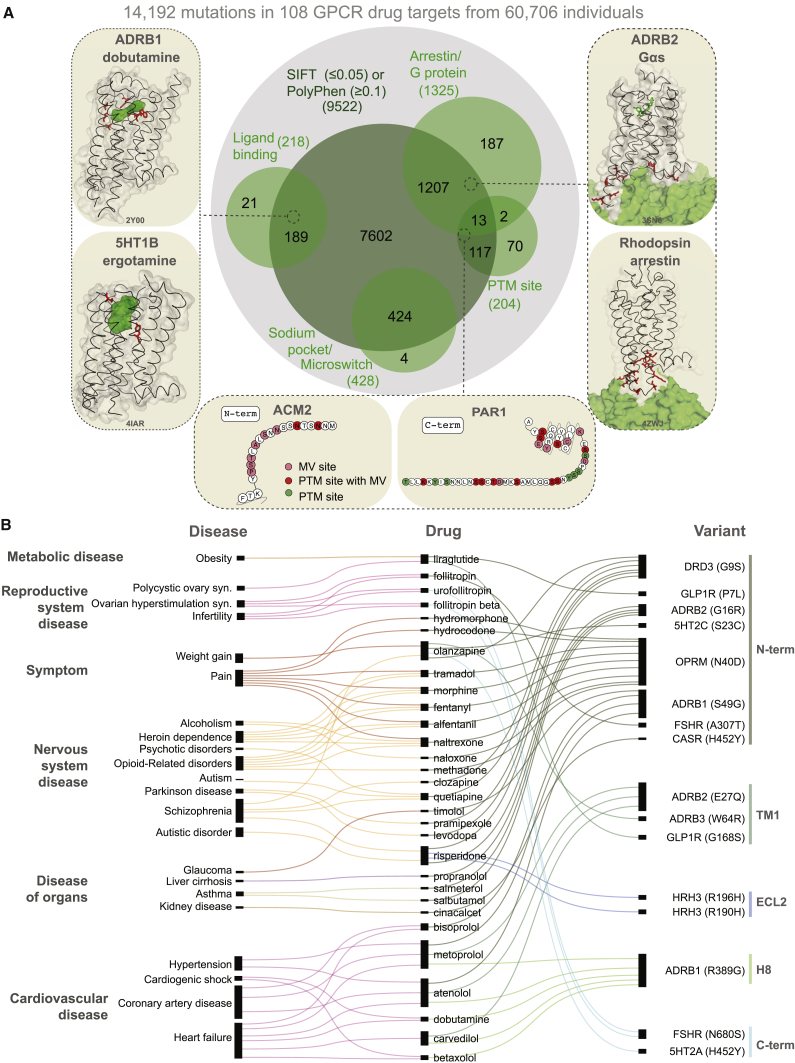
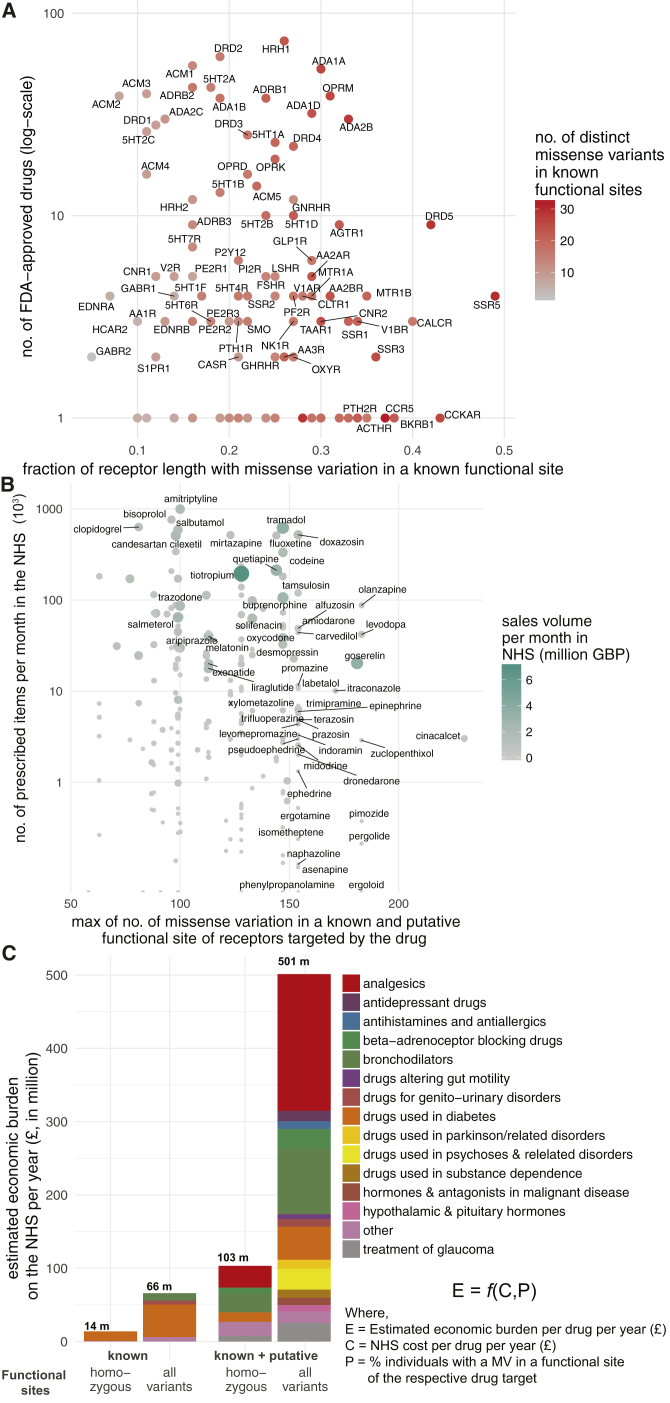
Natural genetic variation in the human genome is a cause of individual differences in responses to medications and is an underappreciated burden on public health. Although 108 G-protein-coupled receptors (GPCRs) are the targets of 475 (∼34%) Food and Drug Administration (FDA)-approved drugs and account for a global sales volume of over 180 billion US dollars annually, the prevalence of genetic variation among GPCRs targeted by drugs is unknown. By analyzing data from 68,496 individuals, we find that GPCRs targeted by drugs show genetic variation within functional regions such as drug- and effector-binding sites in the human population. We experimentally show that certain variants of μ-opioid and Cholecystokinin-A receptors could lead to altered or adverse drug response. By analyzing UK National Health Service drug prescription and sales data, we suggest that characterizing GPCR variants could increase prescription precision, improving patients' quality of life, and relieve the economic and societal burden due to variable drug responsiveness. VIDEO ABSTRACT.
Keywords: FDA approved drugs; GPCR; clinical trial; drug response; economic burden; natural variation; opioid receptor; personalized medicine; pharmacogenomics; polymorphism.
Copyright © 2017 MRC Laboratory of Molecular Biology. Published by Elsevier Inc. All rights reserved.
Figures













Comment in
-
Genetic Vulnerability of GPCRs: A Call to Action.Trends Biochem Sci. 2018 Apr;43(4):227-229. doi: 10.1016/j.tibs.2018.02.006. Trends Biochem Sci. 2018. PMID: 29605062 Free PMC article.
References
-
- Allen J.A., Roth B.L. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011;51:117–144. - PubMed
-
- Boucrot E., Ferreira A.P.A., Almeida-Souza L., Debard S., Vallis Y., Howard G., Bertot L., Sauvonnet N., McMahon H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 2015;517:460–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
