

doi: 10.1016/j.pt.2017.11.007.
Epub 2017 Dec 14.
Tackling Hypotheticals in Helminth Genomes
Affiliations
- PMID: 29249363
- PMCID: PMC11021132
- DOI: 10.1016/j.pt.2017.11.007
Item in Clipboard
Tackling Hypotheticals in Helminth Genomes
Trends Parasitol.
2018 Mar.
Abstract
Advancements in genome sequencing have led to the rapid accumulation of uncharacterized 'hypothetical proteins' in the public databases. Here we provide a community perspective and some best-practice approaches for the accurate functional annotation of uncharacterized genomic sequences.
Keywords: CRISPR; RNAi; annotation; genomes; helminth; hypothetical genes.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures
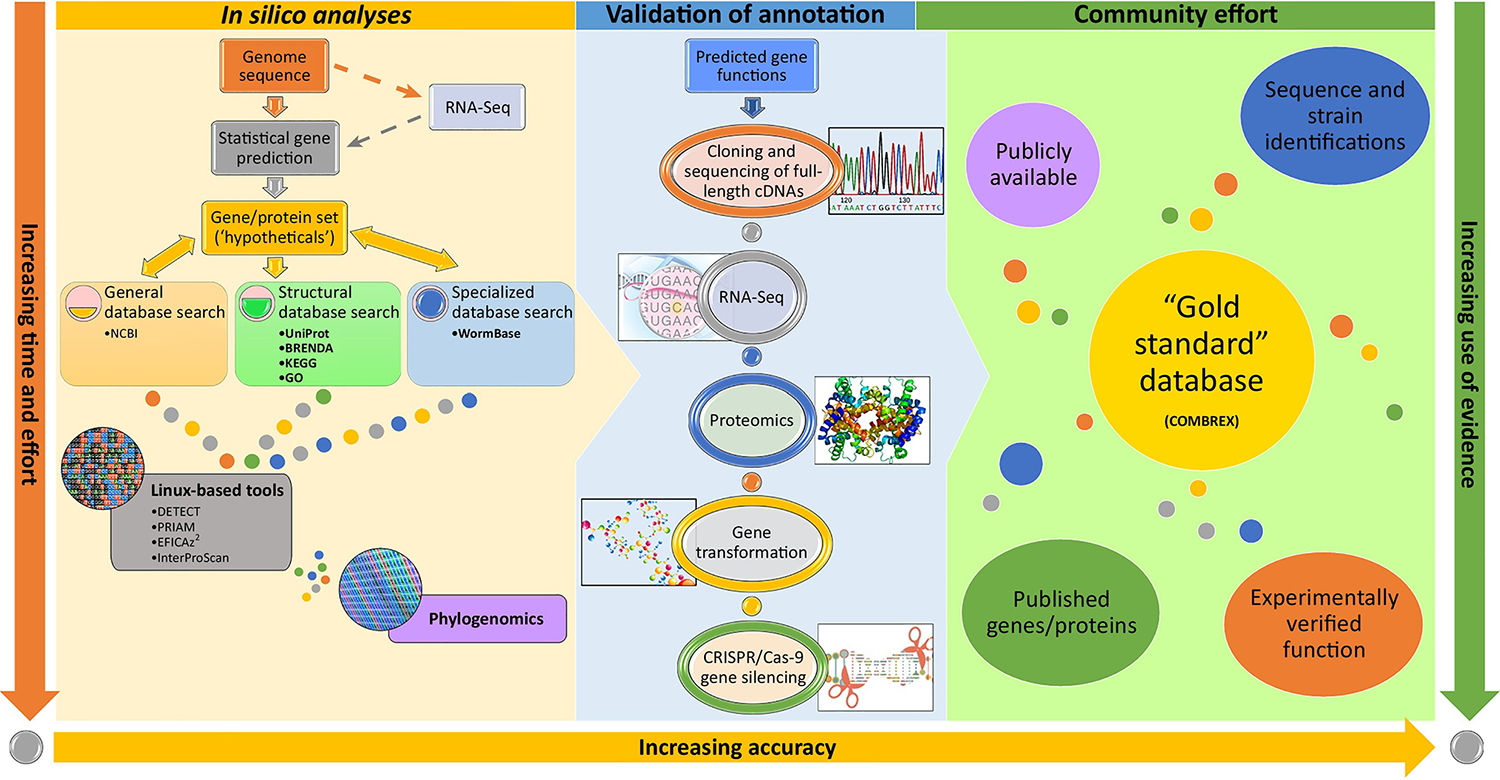
The most efficient means of investigating genes encoded in helminth genomes with the ‘hypothetical’ function annotation is to initially search the currently available sequence databases (typically, NCBI nonredundant database [https://www.ncbi.nlm.nih.gov )] for sequence similarity, using BLAST. This should be followed up by searching structural and specialized databases, for example: protein databases (such as UniProt), enzyme databases (such as BRENDA), and metabolic databases [such as KEGG and Gene Ontology (GO)], for metabolic pathway reconstruction [2]. Several linux-based tools can be used to precisely predict enzyme function, such as DETECT, PRIAM, EFICAz2, and InterProScan. Another in silico method used to improve functional annotation is phylogenomics [3], where hypothetical proteins from phylogenetically related species are compared. Once putative function is determined, cloning and sequencing of full-length cDNAs, proteomics (such as mass spectrometry), and RNA-Seq data can be used to experimentally validate annotations. Additional techniques, such as gene transformation and CRISPR/Cas-9 gene silencing, can also be applied 5, 6, 7, 8, 9, 10. The above mentioned tools and techniques should be used in concert with extensive literature mining to manually curate genomic content. The resulting genes/protein sequences should be deposited in public databases such as COMBREX and WormBase. As the research community accumulates information regarding experimentally verified and published genes/proteins along with species and strain identifications, a ‘Gold Standard’ database can emerge.
References
-
- Leale G, et al. Inferring unknown biological functions by integration of GO annotations and gene expression data. IEEE/ACM Trans. Comput. Biol. Bioinform, 99 (2016), pp. 1–19 arXiv:1608.03672 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
