Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease
- PMID: 29249985
- PMCID: PMC5714892
- DOI: 10.3389/fphys.2017.00982
Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease
Abstract
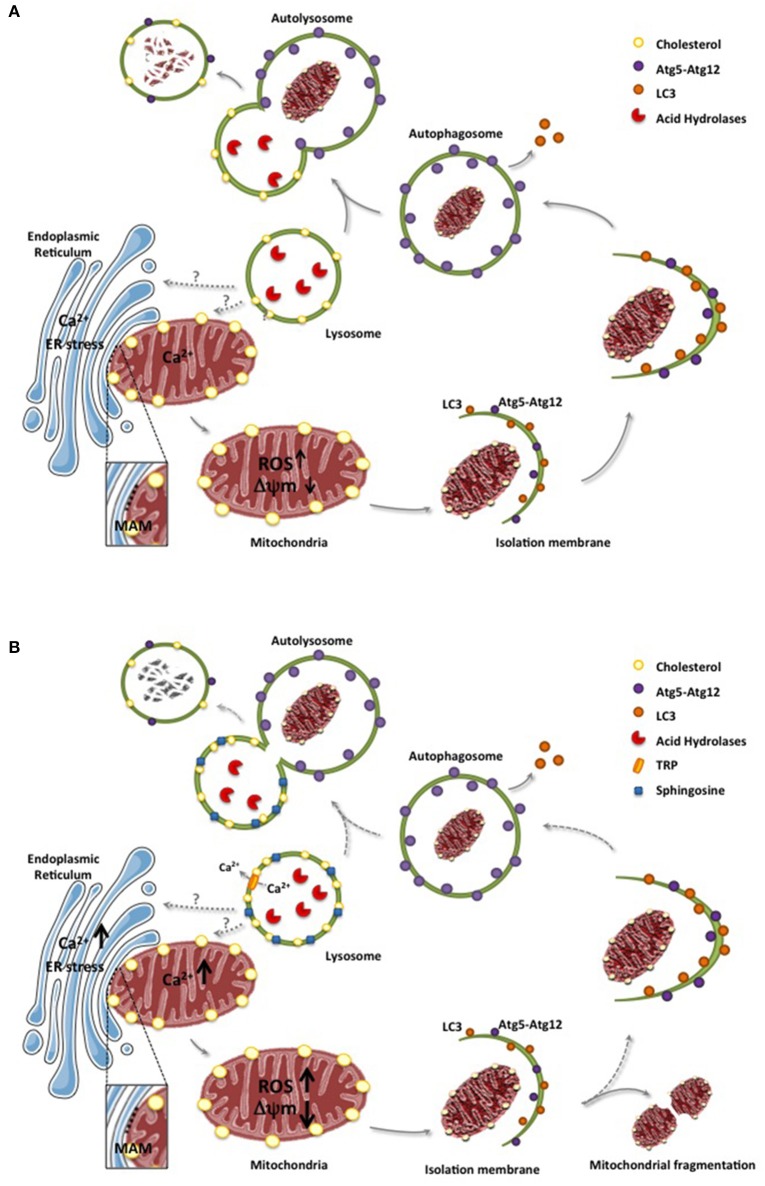
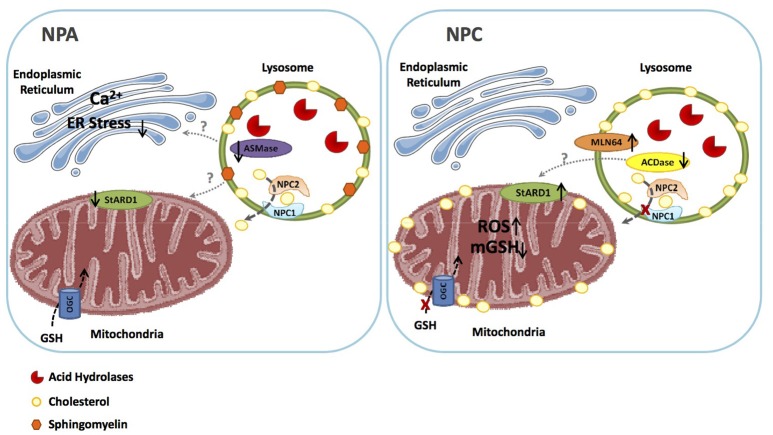
Lysosomal storage disorders (LSD) are characterized by the accumulation of diverse lipid species in lysosomes. Niemann-Pick type A/B (NPA/B) and type C diseases Niemann-Pick type C (NPC) are progressive LSD caused by loss of function of distinct lysosomal-residing proteins, acid sphingomyelinase and NPC1, respectively. While the primary cause of these diseases differs, both share common biochemical features, including the accumulation of sphingolipids and cholesterol, predominantly in endolysosomes. Besides these alterations in lysosomal homeostasis and function due to accumulation of specific lipid species, the lysosomal functional defects can have far-reaching consequences, disrupting intracellular trafficking of sterols, lipids and calcium through membrane contact sites (MCS) of apposed compartments. Although MCS between endoplasmic reticulum and mitochondria have been well studied and characterized in different contexts, emerging evidence indicates that lysosomes also exhibit close proximity with mitochondria, which translates in their mutual functional regulation. Indeed, as best illustrated in NPC disease, alterations in the lysosomal-mitochondrial liaisons underlie the secondary accumulation of specific lipids, such as cholesterol in mitochondria, resulting in mitochondrial dysfunction and defective antioxidant defense, which contribute to disease progression. Thus, a better understanding of the lysosomal and mitochondrial interactions and trafficking may identify novel targets for the treatment of Niemann-Pick disease.
Keywords: acid sphingomyelinase; cholesterol; intracellular trafficking; lysosomal disorders; lysosomes; mitochondria; sphingolipids.
Figures