

High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation
- PMID: 29253866
- PMCID: PMC5734774
- DOI: 10.1371/journal.pone.0189489
High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation
Abstract
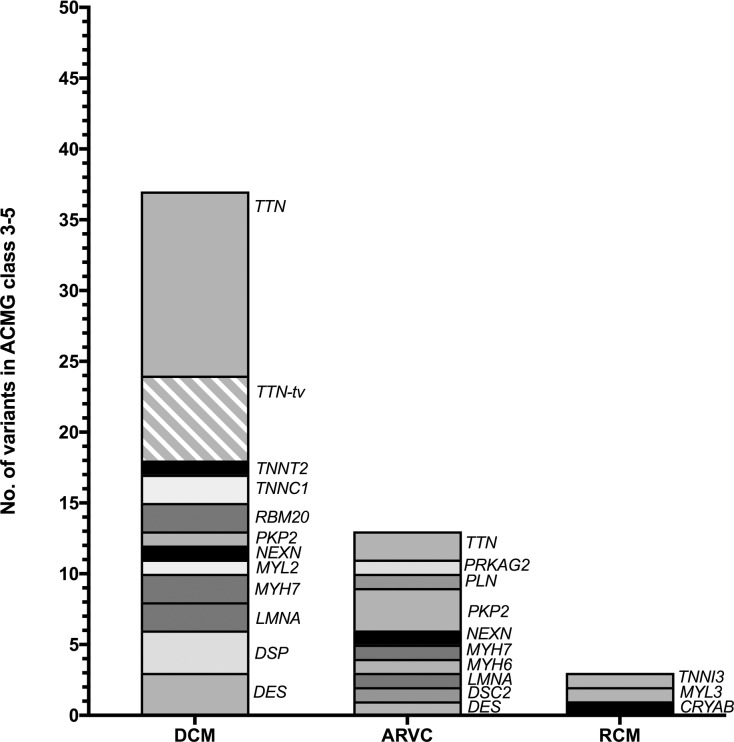
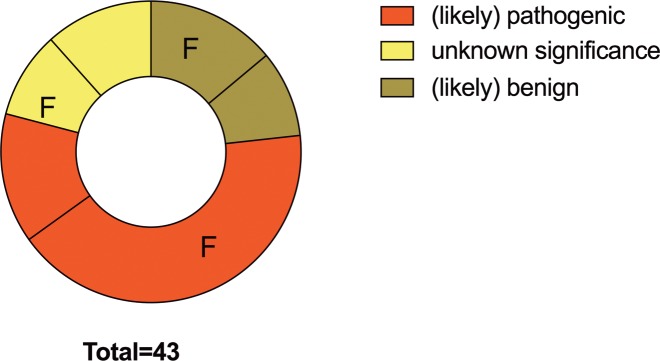
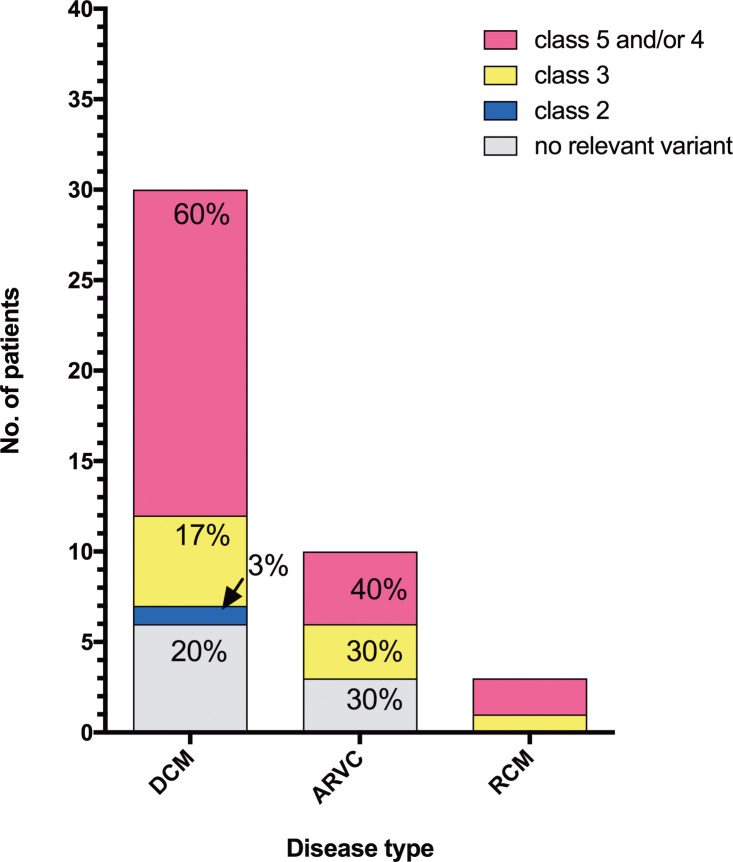
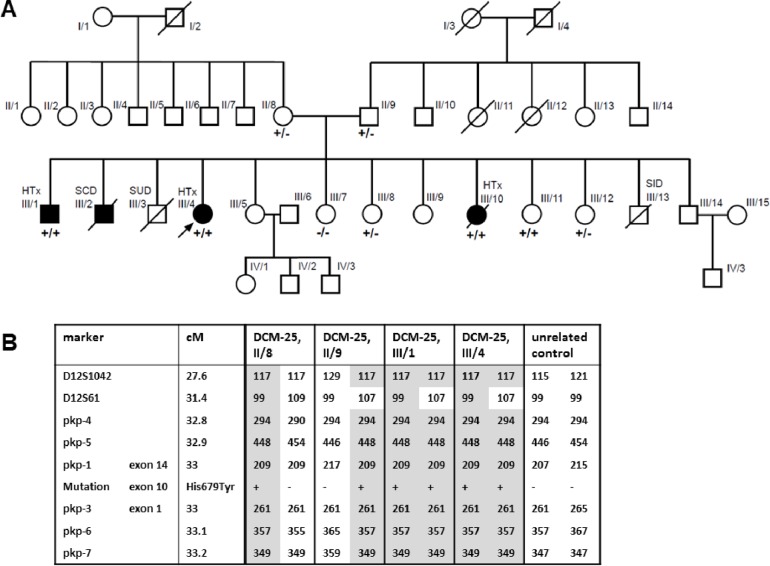
Cardiomyopathies might lead to end-stage heart disease with the requirement of drastic treatments like bridging up to transplant or heart transplantation. A not precisely known proportion of these diseases are genetically determined. We genotyped 43 index-patients (30 DCM, 10 ARVC, 3 RCM) with advanced or end stage cardiomyopathy using a gene panel which covered 46 known cardiomyopathy disease genes. Fifty-three variants with possible impact on disease in 33 patients were identified. Of these 27 (51%) were classified as likely pathogenic or pathogenic in the MYH7, MYL2, MYL3, NEXN, TNNC1, TNNI3, DES, LMNA, PKP2, PLN, RBM20, TTN, and CRYAB genes. Fifty-six percent (n = 24) of index-patients carried a likely pathogenic or pathogenic mutation. Of these 75% (n = 18) were familial and 25% (n = 6) sporadic cases. However, severe cardiomyopathy seemed to be not characterized by a specific mutation profile. Remarkably, we identified a novel homozygous PKP2-missense variant in a large consanguineous family with sudden death in early childhood and several members with heart transplantation in adolescent age.
Conflict of interest statement
Figures
References
-
- Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. 2013;6(4):337–46. Epub 2013/07/19. doi: 10.1161/CIRCGENETICS.113.000039 CIRCGENETICS.113.000039 [pii]. ; PubMed Central PMCID: PMC3887521. - DOI - PMC - PubMed
-
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16(8):601–8. Epub 2014/02/08. doi: 10.1038/gim.2013.204 gim2013204 [pii]. . - DOI - PubMed
-
- Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, et al. Targeted Analysis of Whole Genome Sequence Data to Diagnose Genetic Cardiomyopathy. Circulation: Cardiovascular Genetics. 2014;7(6):751–9. doi: 10.1161/circgenetics.113.000578 - DOI - PMC - PubMed
-
- Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2014. Epub 2014/08/29. doi: ehu301 [pii] doi: 10.1093/eurheartj/ehu301 . - DOI - PubMed
-
- van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years' experience. Eur J Heart Fail. 2013;15(6):628–36. Epub 2013/01/26. doi: 10.1093/eurjhf/hft013 hft013 [pii]. . - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
