

N-Glycan-dependent protein folding and endoplasmic reticulum retention regulate GPI-anchor processing
- PMID: 29255114
- PMCID: PMC5800811
- DOI: 10.1083/jcb.201706135
N-Glycan-dependent protein folding and endoplasmic reticulum retention regulate GPI-anchor processing
Abstract
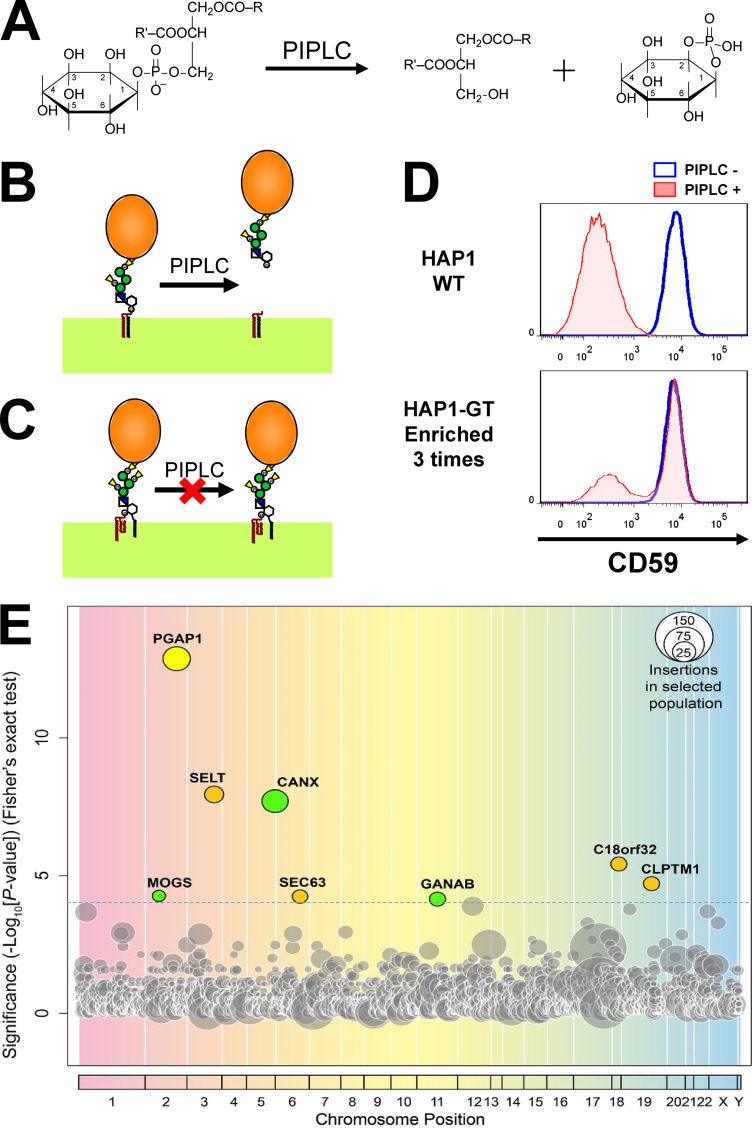
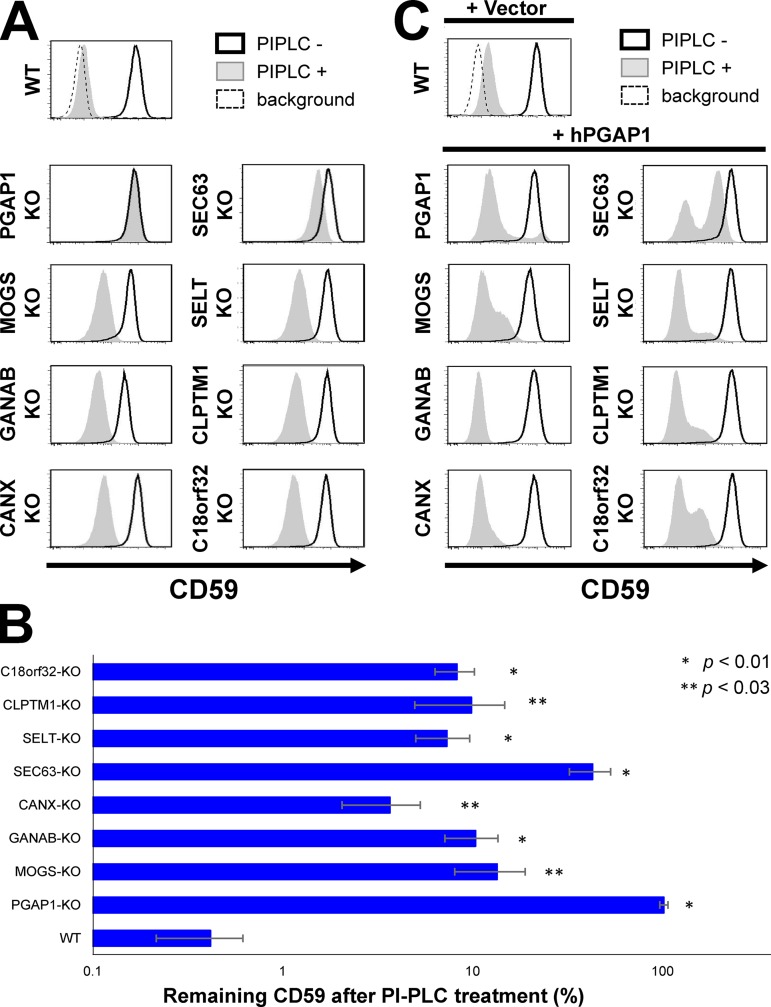
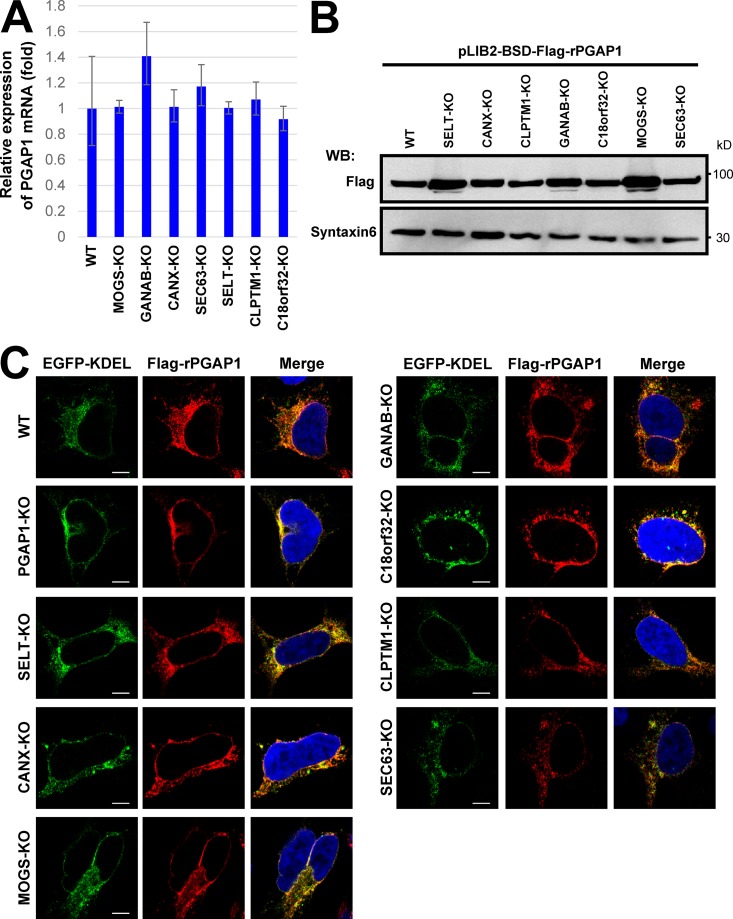
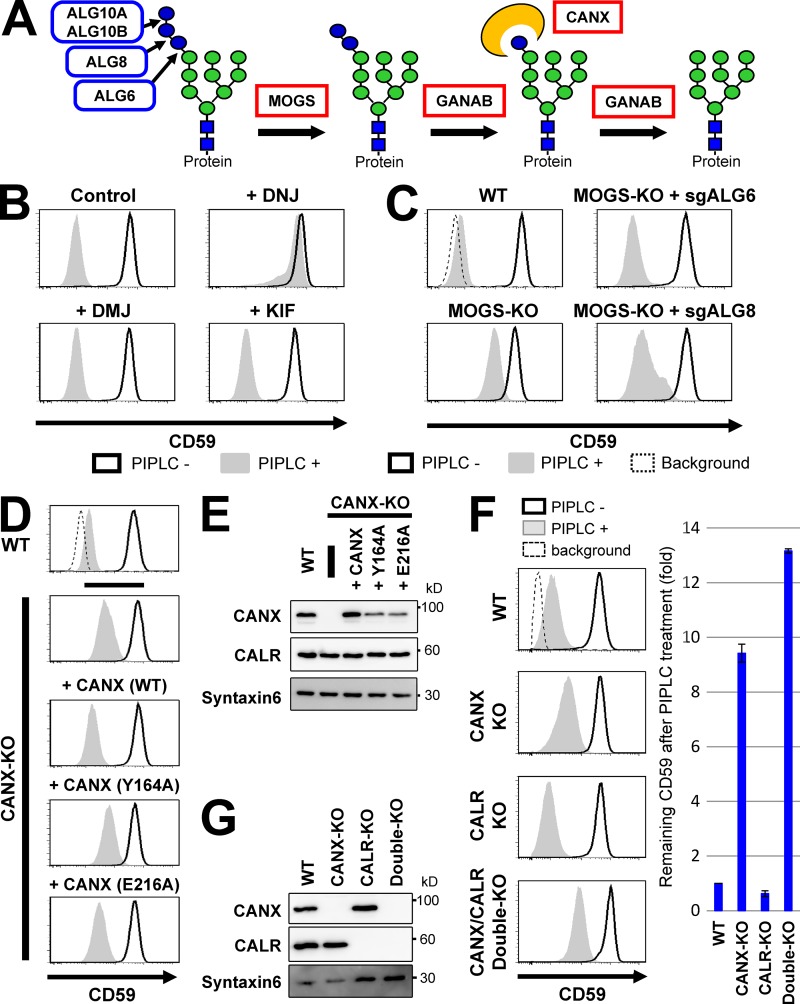
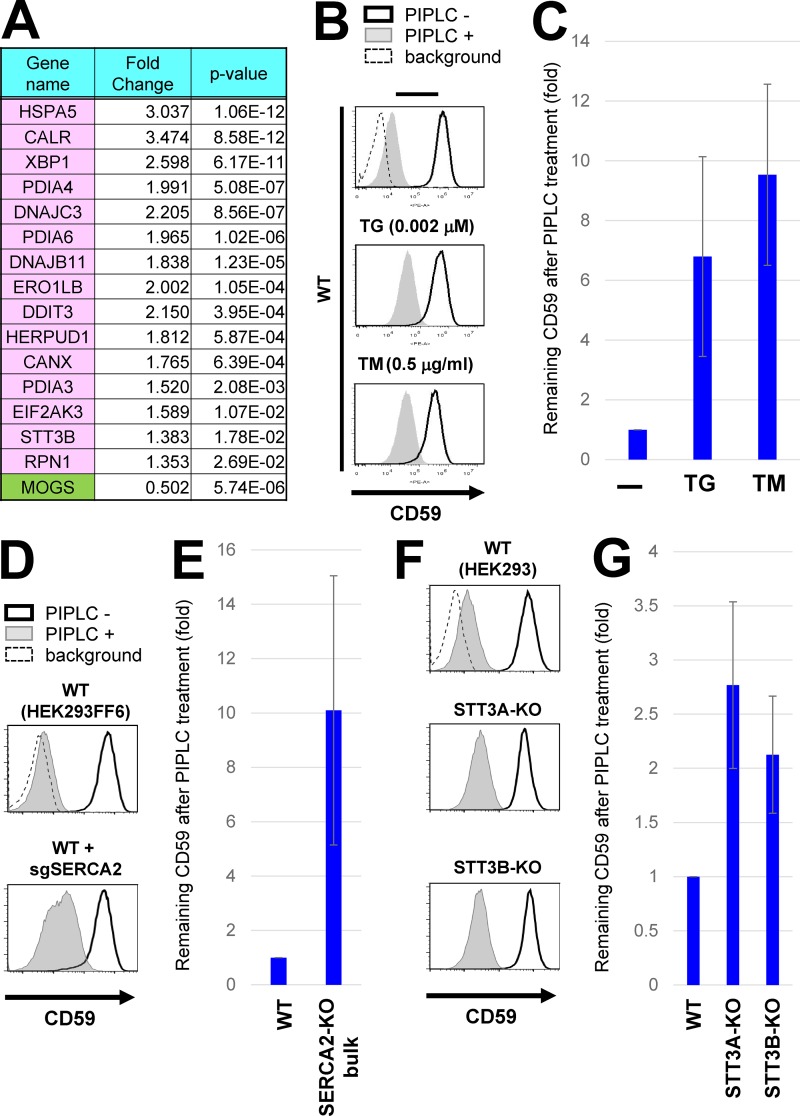
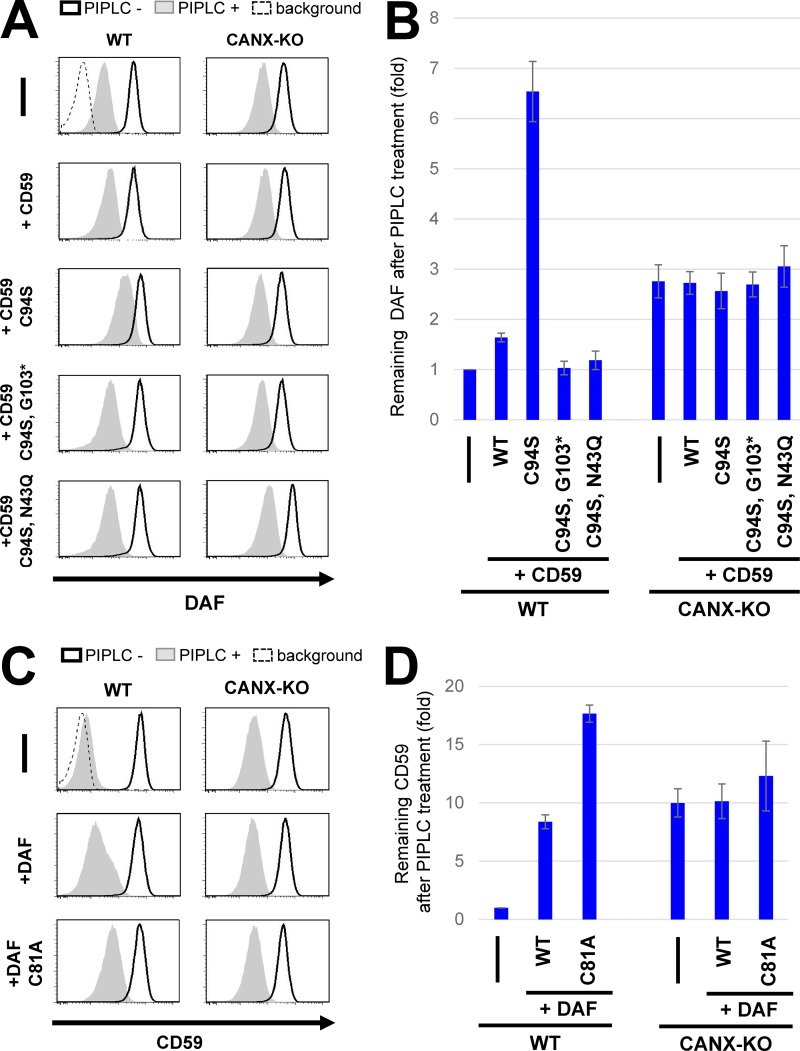
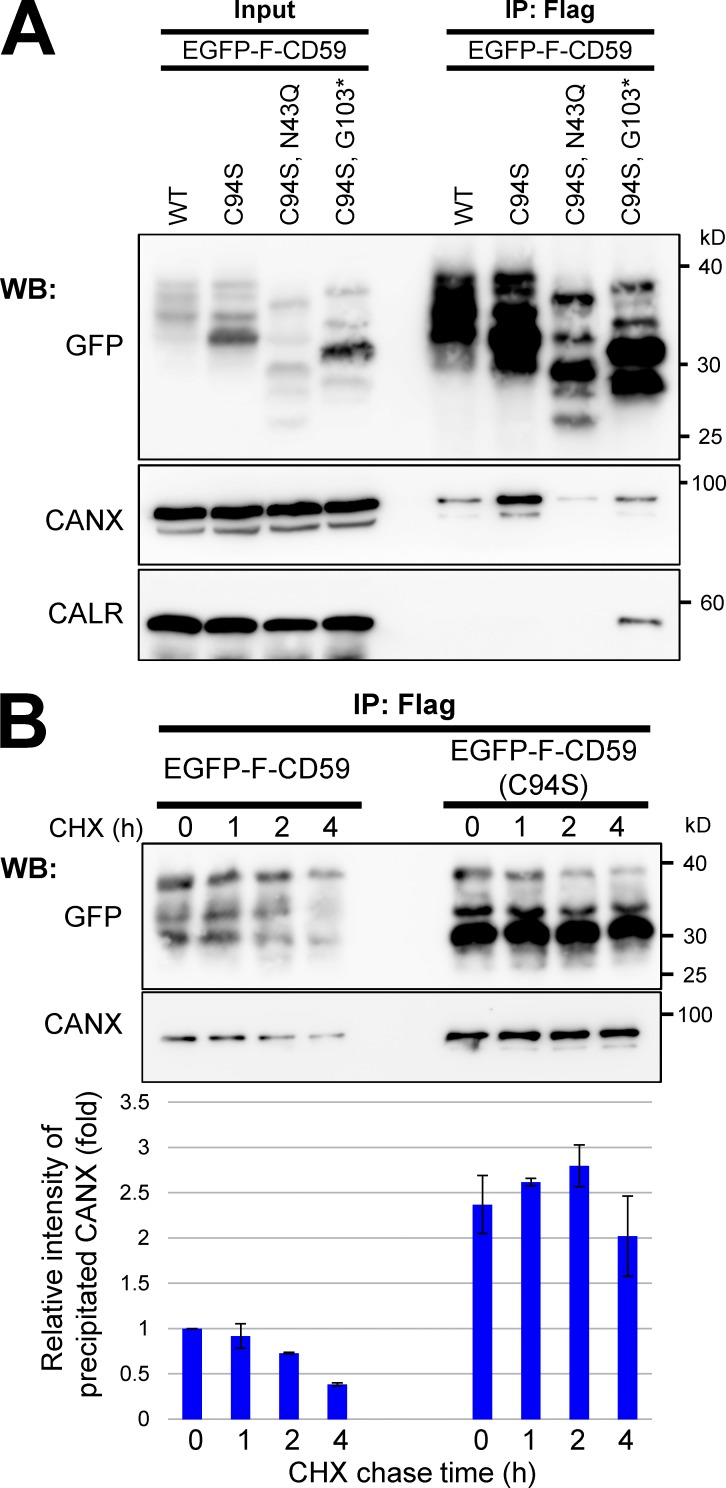
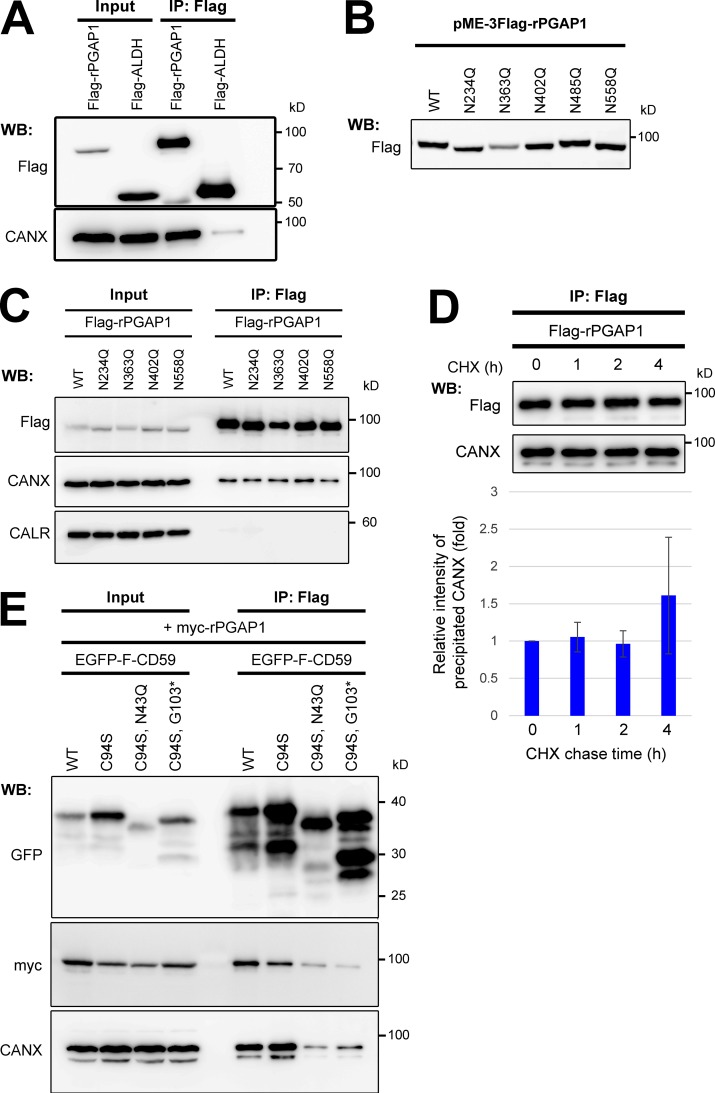
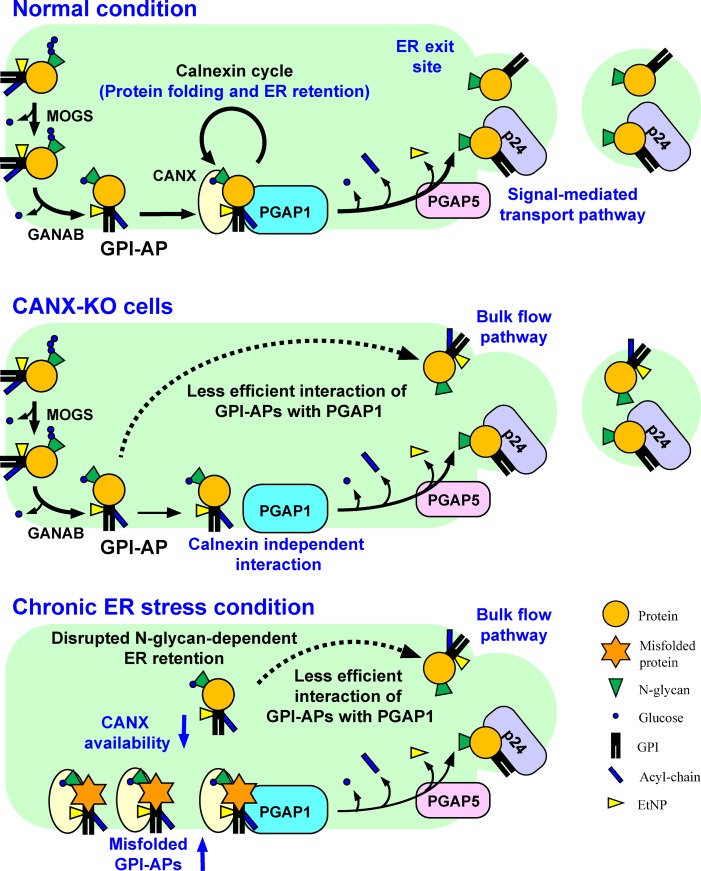
Glycosylphosphatidylinositol (GPI) anchoring of proteins is a conserved posttranslational modification in the endoplasmic reticulum (ER). Soon after GPI is attached, an acyl chain on the GPI inositol is removed by post-GPI attachment to proteins 1 (PGAP1), a GPI-inositol deacylase. This is crucial for switching GPI-anchored proteins (GPI-APs) from protein folding to transport states. We performed haploid genetic screens to identify factors regulating GPI-inositol deacylation, identifying seven genes. In particular, calnexin cycle impairment caused inefficient GPI-inositol deacylation. Calnexin was specifically associated with GPI-APs, dependent on N-glycan and GPI moieties, and assisted efficient GPI-inositol deacylation by PGAP1. Under chronic ER stress caused by misfolded GPI-APs, inositol-acylated GPI-APs were exposed on the cell surface. These results indicated that N-glycans participate in quality control and temporal ER retention of GPI-APs, ensuring their correct folding and GPI processing before exiting from the ER. Once the system is disrupted by ER stress, unprocessed GPI-APs become exposed on the cell surface.
© 2018 Liu et al.
Figures
Similar articles
-
Calnexin mediates the maturation of GPI-anchors through ER retention.J Biol Chem. 2020 Nov 27;295(48):16393-16410. doi: 10.1074/jbc.RA120.015577. Epub 2020 Sep 23. J Biol Chem. 2020. PMID: 32967966 Free PMC article.
-
Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p.J Biol Chem. 2004 Apr 2;279(14):14256-63. doi: 10.1074/jbc.M313755200. Epub 2004 Jan 20. J Biol Chem. 2004. PMID: 14734546
-
AtPGAP1 functions as a GPI inositol-deacylase required for efficient transport of GPI-anchored proteins.Plant Physiol. 2021 Dec 4;187(4):2156-2173. doi: 10.1093/plphys/kiab384. Plant Physiol. 2021. PMID: 34618080 Free PMC article.
-
Trafficking of glycosylphosphatidylinositol anchored proteins from the endoplasmic reticulum to the cell surface.J Lipid Res. 2016 Mar;57(3):352-60. doi: 10.1194/jlr.R062760. Epub 2015 Oct 8. J Lipid Res. 2016. PMID: 26450970 Free PMC article. Review.
-
Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins.Prog Lipid Res. 2011 Oct;50(4):411-24. doi: 10.1016/j.plipres.2011.05.002. Epub 2011 May 30. Prog Lipid Res. 2011. PMID: 21658410 Review.
Cited by
-
Molecular basis of the inositol deacylase PGAP1 involved in quality control of GPI-AP biogenesis.Nat Commun. 2024 Jan 2;15(1):8. doi: 10.1038/s41467-023-44568-2. Nat Commun. 2024. PMID: 38167496 Free PMC article.
-
Differential expression of miRNAs and functional role of mir-200a in high and low productivity CHO cells expressing an Fc fusion protein.Biotechnol Lett. 2021 Aug;43(8):1551-1563. doi: 10.1007/s10529-021-03153-7. Epub 2021 Jun 16. Biotechnol Lett. 2021. PMID: 34131805 Free PMC article.
-
Calnexin mediates the maturation of GPI-anchors through ER retention.J Biol Chem. 2020 Nov 27;295(48):16393-16410. doi: 10.1074/jbc.RA120.015577. Epub 2020 Sep 23. J Biol Chem. 2020. PMID: 32967966 Free PMC article.
-
Endoplasmic Reticulum Export of GPI-Anchored Proteins.Int J Mol Sci. 2019 Jul 17;20(14):3506. doi: 10.3390/ijms20143506. Int J Mol Sci. 2019. PMID: 31319476 Free PMC article. Review.
-
The Glycosylphosphatidylinositol Anchor: A Linchpin for Cell Surface Versatility of Trypanosomatids.Front Cell Dev Biol. 2021 Nov 1;9:720536. doi: 10.3389/fcell.2021.720536. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34790656 Free PMC article. Review.
References
-
- Boukhzar L., Hamieh A., Cartier D., Tanguy Y., Alsharif I., Castex M., Arabo A., El Hajji S., Bonnet J.J., Errami M., et al. . 2016. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 24:557–574. 10.1089/ars.2015.6478 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
