

Novel inhibitors targeting Venezuelan equine encephalitis virus capsid protein identified using In Silico Structure-Based-Drug-Design
- PMID: 29255256
- PMCID: PMC5735092
- DOI: 10.1038/s41598-017-17672-9
Novel inhibitors targeting Venezuelan equine encephalitis virus capsid protein identified using In Silico Structure-Based-Drug-Design
Erratum in
-
Author Correction: Novel inhibitors targeting Venezuelan equine encephalitis virus capsid protein identified using In Silico Structure-Based-Drug-Design.Sci Rep. 2018 Sep 4;8(1):13487. doi: 10.1038/s41598-018-31644-7. Sci Rep. 2018. PMID: 30177745 Free PMC article.
Abstract
Therapeutics are currently unavailable for Venezuelan equine encephalitis virus (VEEV), which elicits flu-like symptoms and encephalitis in humans, with an estimated 14% of cases resulting in neurological disease. Here we identify anti-VEEV agents using in silico structure-based-drug-design (SBDD) for the first time, characterising inhibitors that block recognition of VEEV capsid protein (C) by the host importin (IMP) α/β1 nuclear transport proteins. From an initial screen of 1.5 million compounds, followed by in silico refinement and screening for biological activity in vitro, we identified 21 hit compounds which inhibited IMPα/β1:C binding with IC50s as low as 5 µM. Four compounds were found to inhibit nuclear import of C in transfected cells, with one able to reduce VEEV replication at µM concentration, concomitant with reduced C nuclear accumulation in infected cells. Further, this compound was inactive against a mutant VEEV that lacks high affinity IMPα/β1:C interaction, supporting the mode of its antiviral action to be through inhibiting C nuclear localization. This successful application of SBDD paves the way for lead optimization for VEEV antivirals, and is an exciting prospect to identify inhibitors for the many other viral pathogens of significance that require IMPα/β1 in their infectious cycle.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
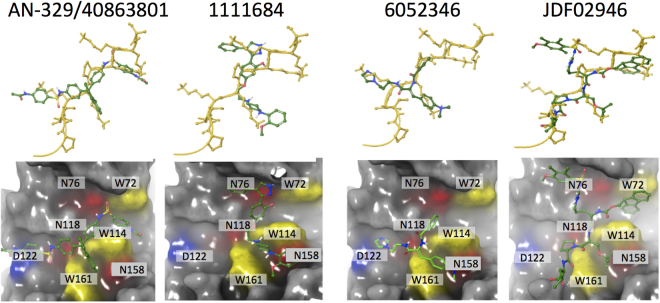
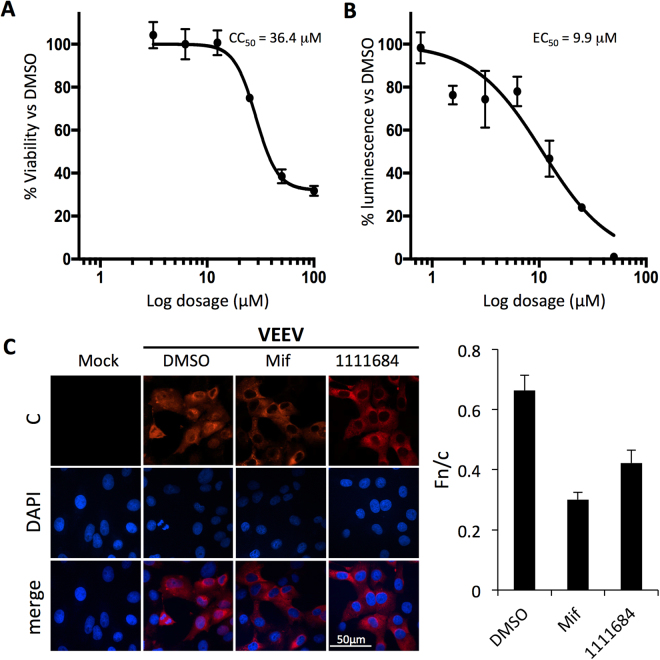
References
-
- Frolov, I. Persistent infection and suppression of host response by alphaviruses. Archives of virology. Supplementum, 139–147 (2004). - PubMed
-
- Griffin, D. E. In Fields’ Virology Vol. 1 (eds D. M. Knipe & P. M. Howley) Ch. 31, 1023–1069 (Wolters Kluwer Health/Lippincott Williams & Wilkins, 2007).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
