

Severe infections emerge from commensal bacteria by adaptive evolution
- PMID: 29256859
- PMCID: PMC5736351
- DOI: 10.7554/eLife.30637
Severe infections emerge from commensal bacteria by adaptive evolution
Abstract
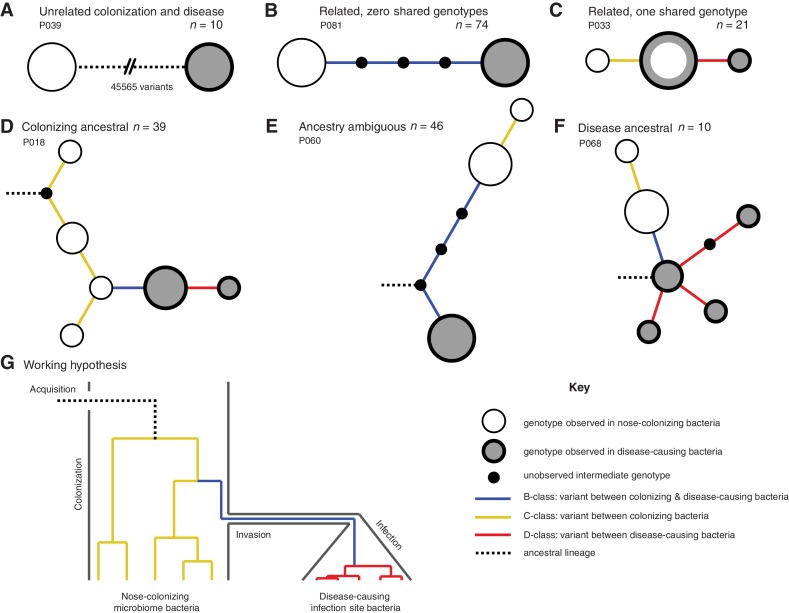
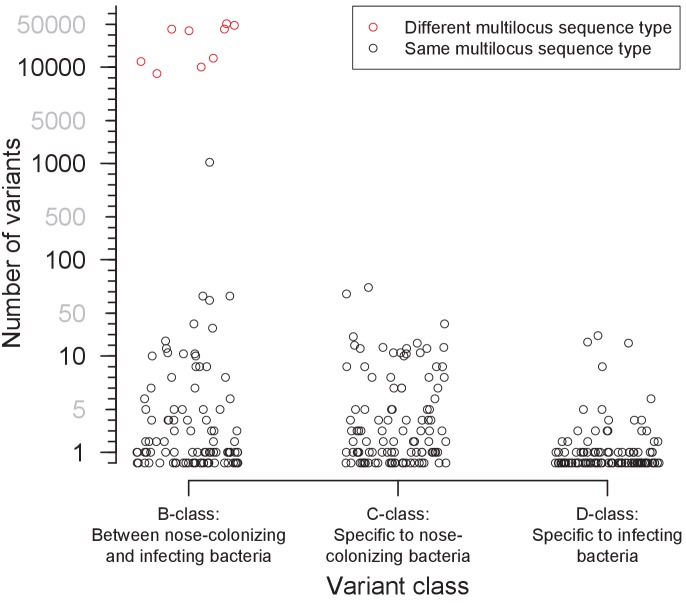
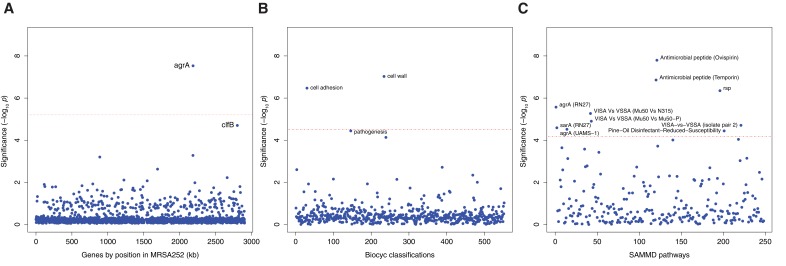
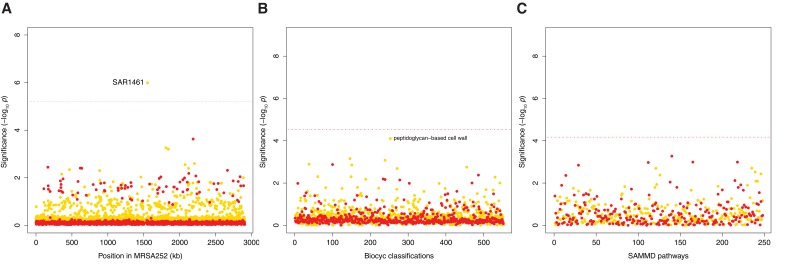
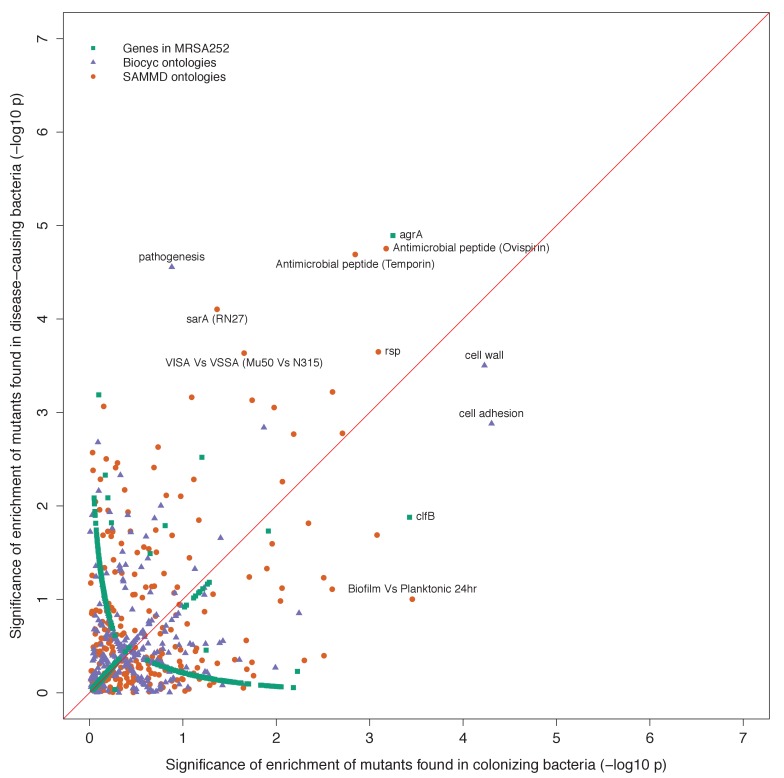
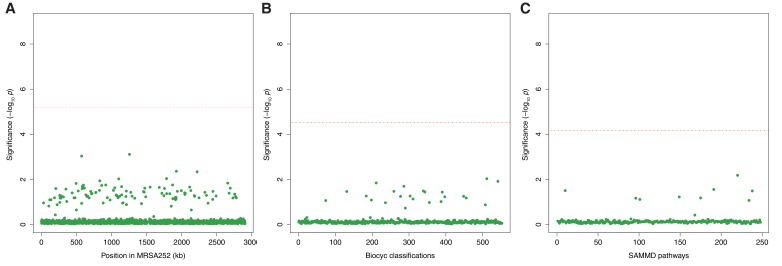
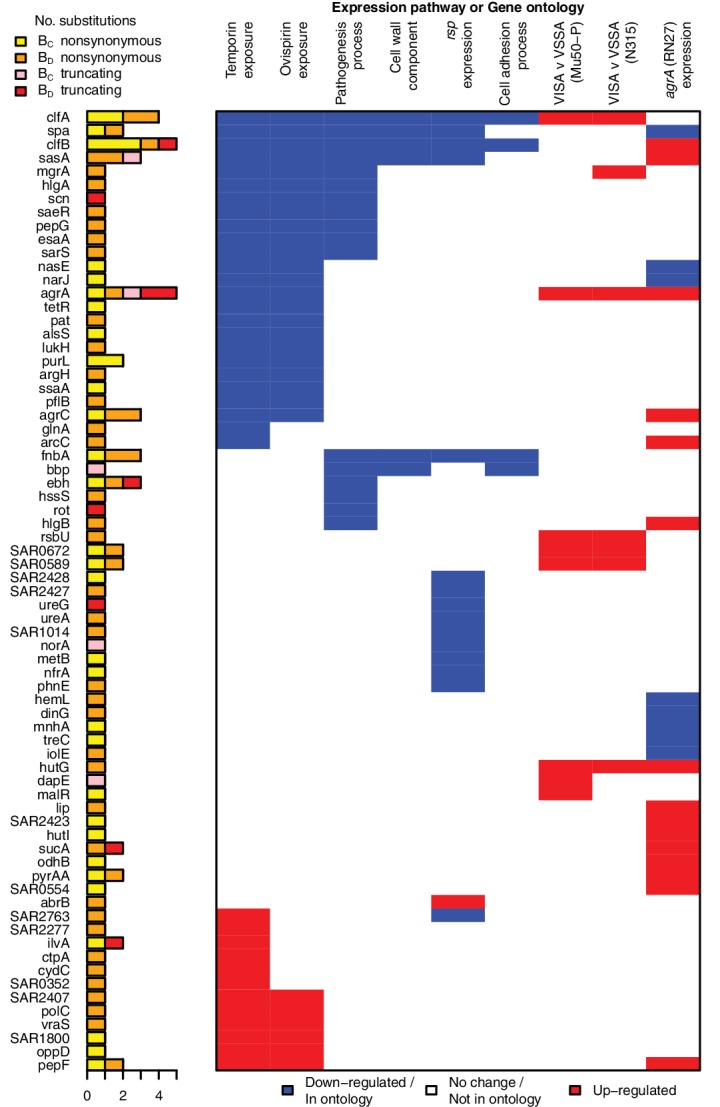
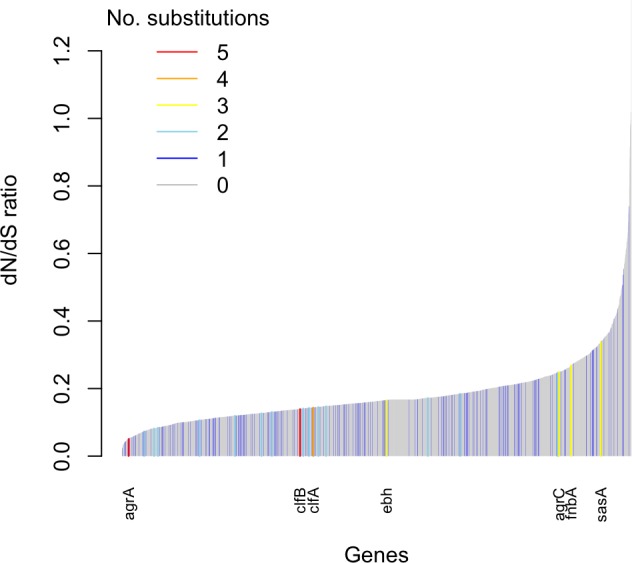
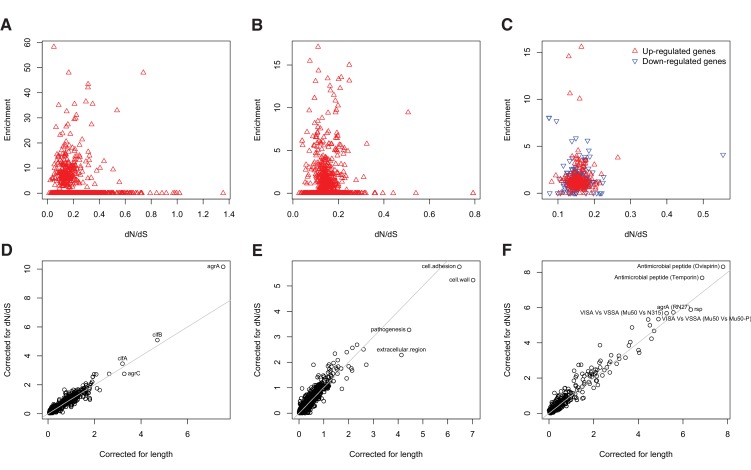
Bacteria responsible for the greatest global mortality colonize the human microbiota far more frequently than they cause severe infections. Whether mutation and selection among commensal bacteria are associated with infection is unknown. We investigated de novo mutation in 1163 Staphylococcus aureus genomes from 105 infected patients with nose colonization. We report that 72% of infections emerged from the nose, with infecting and nose-colonizing bacteria showing parallel adaptive differences. We found 2.8-to-3.6-fold adaptive enrichments of protein-altering variants in genes responding to rsp, which regulates surface antigens and toxin production; agr, which regulates quorum-sensing, toxin production and abscess formation; and host-derived antimicrobial peptides. Adaptive mutations in pathogenesis-associated genes were 3.1-fold enriched in infecting but not nose-colonizing bacteria. None of these signatures were observed in healthy carriers nor at the species-level, suggesting infection-associated, short-term, within-host selection pressures. Our results show that signatures of spontaneous adaptive evolution are specifically associated with infection, raising new possibilities for diagnosis and treatment.
Keywords: Staphylococcus aureus; adaptation; human; infection; infectious disease; microbiology; pathogen genomics; virulence; within-host evolution.
Conflict of interest statement
No competing interests declared.
Figures
References
-
- Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. Journal of Bacteriology. 2008;190:300–310. doi: 10.1128/JB.01000-07. - DOI - PMC - PubMed
-
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research. 2016;44:D471–D480. doi: 10.1093/nar/gkv1164. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
