

ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules
- PMID: 29257127
- PMCID: PMC5735918
- DOI: 10.1038/sdata.2017.193
ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules
Abstract
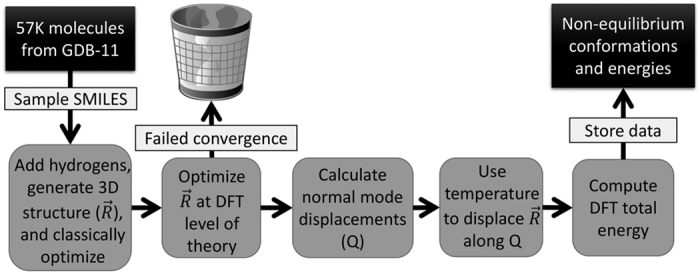
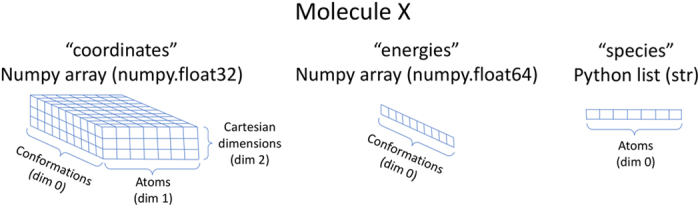
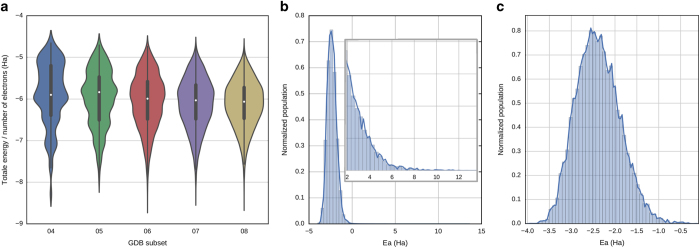
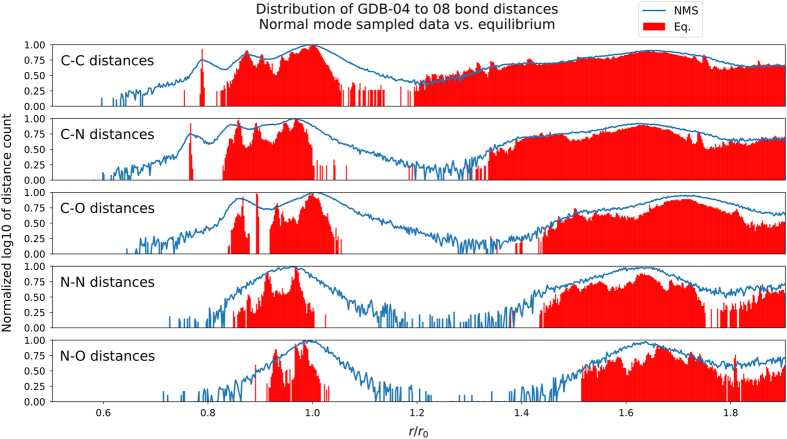
One of the grand challenges in modern theoretical chemistry is designing and implementing approximations that expedite ab initio methods without loss of accuracy. Machine learning (ML) methods are emerging as a powerful approach to constructing various forms of transferable atomistic potentials. They have been successfully applied in a variety of applications in chemistry, biology, catalysis, and solid-state physics. However, these models are heavily dependent on the quality and quantity of data used in their fitting. Fitting highly flexible ML potentials, such as neural networks, comes at a cost: a vast amount of reference data is required to properly train these models. We address this need by providing access to a large computational DFT database, which consists of more than 20 M off equilibrium conformations for 57,462 small organic molecules. We believe it will become a new standard benchmark for comparison of current and future methods in the ML potential community.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Dataset use reported in
- doi: 10.1039/C6SC05720A
References
Data Citations
-
- Smith J. S., Isayev O., Roitberg A. E. 2017. Figshare. https://doi.org/10.6084/m9.figshare.c.3846712 - DOI
References
-
- Becke A. D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 140, 18A301 (2014). - PubMed
-
- Grimme S., Antony J., Schwabe T. & Mück-Lichtenfeld C. Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of (bio)organic molecules. Org. Biomol. Chem. 5, 741–758 (2007). - PubMed
-
- te Velde G. et al. Chemistry with ADF. J. Comput. Chem. 22, 931–967 (2001).
-
- Brunk E. & Rothlisberger U. Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States. Chemical Reviews 115, 6217–6263 (2015). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
