

KBG syndrome
- PMID: 29258554
- PMCID: PMC5735576
- DOI: 10.1186/s13023-017-0736-8
KBG syndrome
Abstract
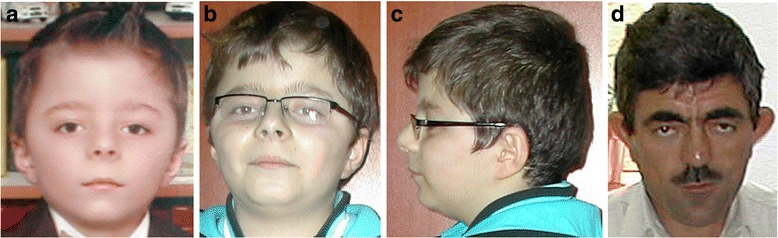
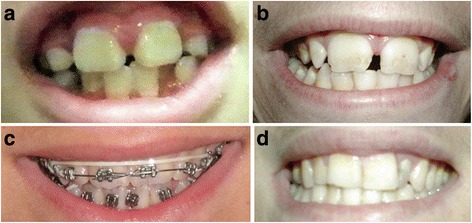
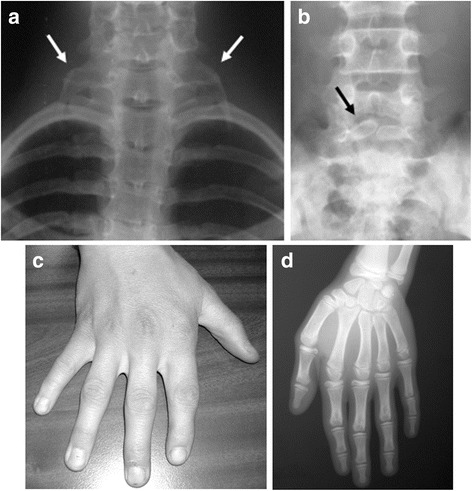
Clinical description: KBG syndrome is characterized by macrodontia of upper central incisors, distinctive craniofacial features such as triangular face, prominent nasal bridge, thin upper lip and synophrys; skeletal findings including short stature, delayed bone age, and costovertebral anomalies; and developmental delay/intellectual disability sometimes associated with seizures and EEG abnormalities. The condition was named KBG syndrome after the initials of the last names of three original families reported in 1975.
Epidemiology: The prevalence of KBG syndrome is not established. There are over 100 patients reported in the literature. It is likely that KBG syndrome is underreported due to incomplete recognition and very mild presentations of the disorder in some individuals. KBG syndrome is typically milder in females.
Etiology: Causative variants in ANKRD11 have been identified in affected individuals. The vast majority of identified variants are loss of function, which include nonsense and frameshift variants and larger deletions at 16q24.3. Haploinsufficiency appears to be the mechanism of pathogenicity.
Genetic counseling: Familial and de novo cases have been reported. Causative de novo variants occur approximately one third of the time. Transmission follows an autosomal dominant pattern. The syndrome displays inter- and intra-familial variability.
Keywords: ANKRD11; KBG syndrome; Macrodontia; Review; Short stature.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patients or their guardian/parent/next of kin for the publication of this report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
