

Evolutionary expansion of DNA hypomethylation in the mammalian germline genome
- PMID: 29259021
- PMCID: PMC5793779
- DOI: 10.1101/gr.225896.117
Evolutionary expansion of DNA hypomethylation in the mammalian germline genome
Erratum in
-
Corrigendum: Evolutionary expansion of DNA hypomethylation in the mammalian germline genome.Genome Res. 2018 May;28(5):766.2. doi: 10.1101/gr.236885.118. Genome Res. 2018. PMID: 29717002 Free PMC article. No abstract available.
Abstract
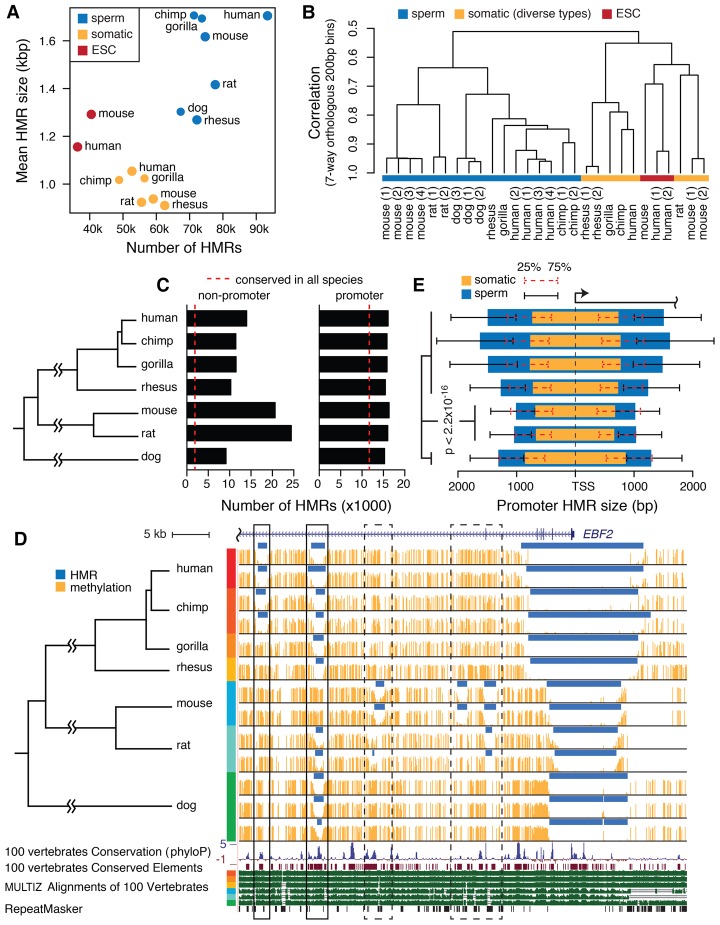
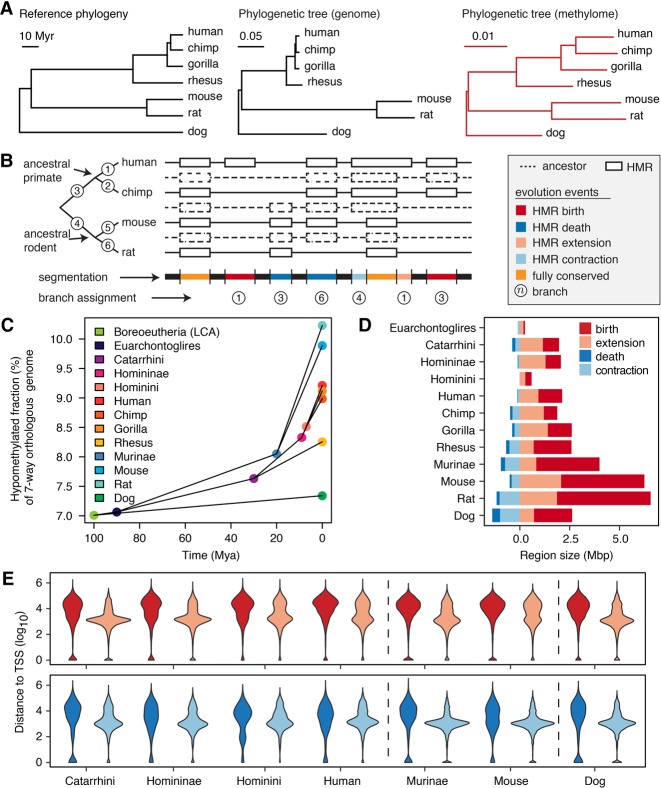
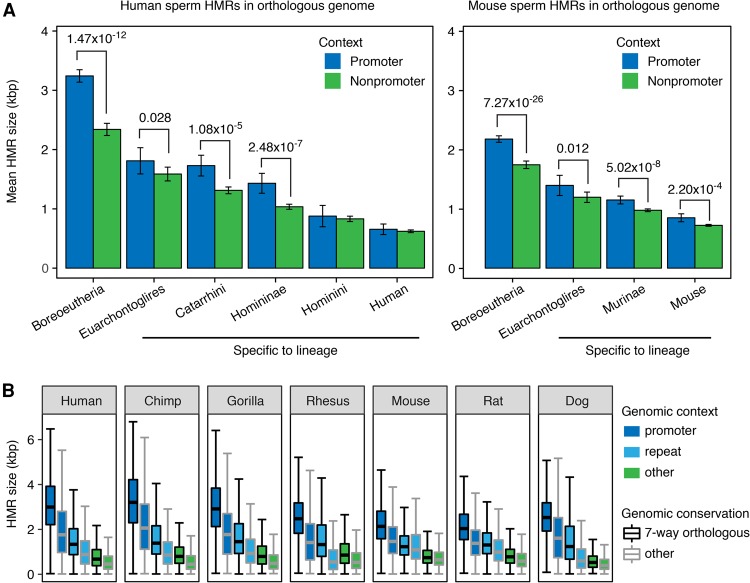
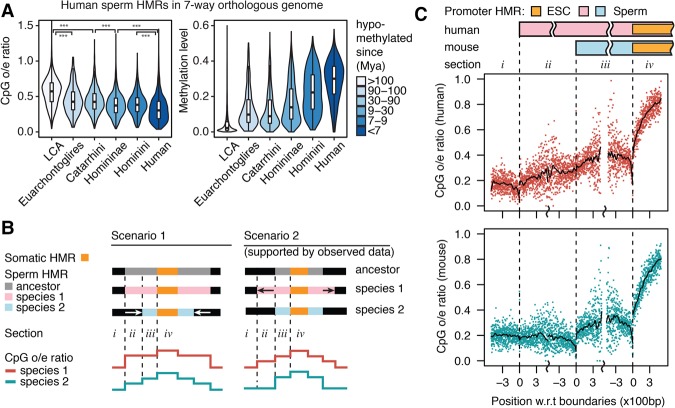
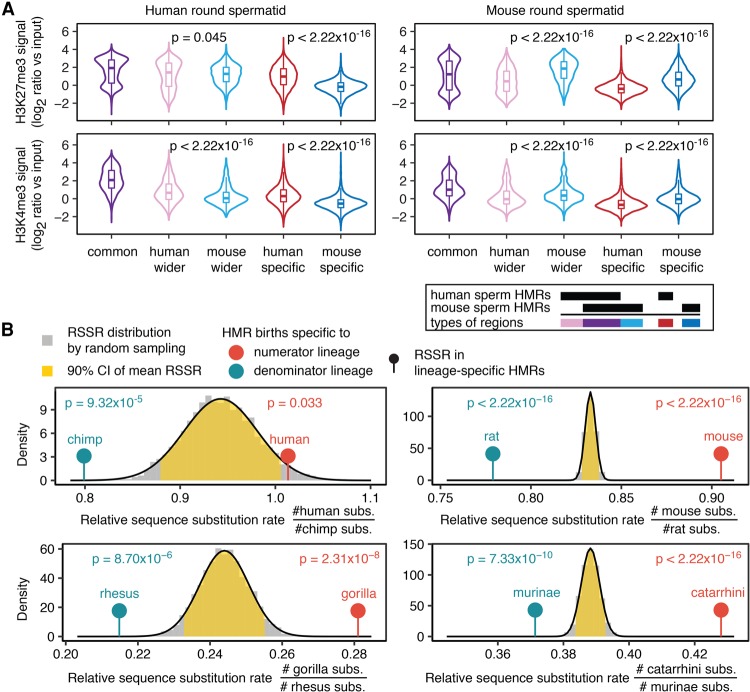
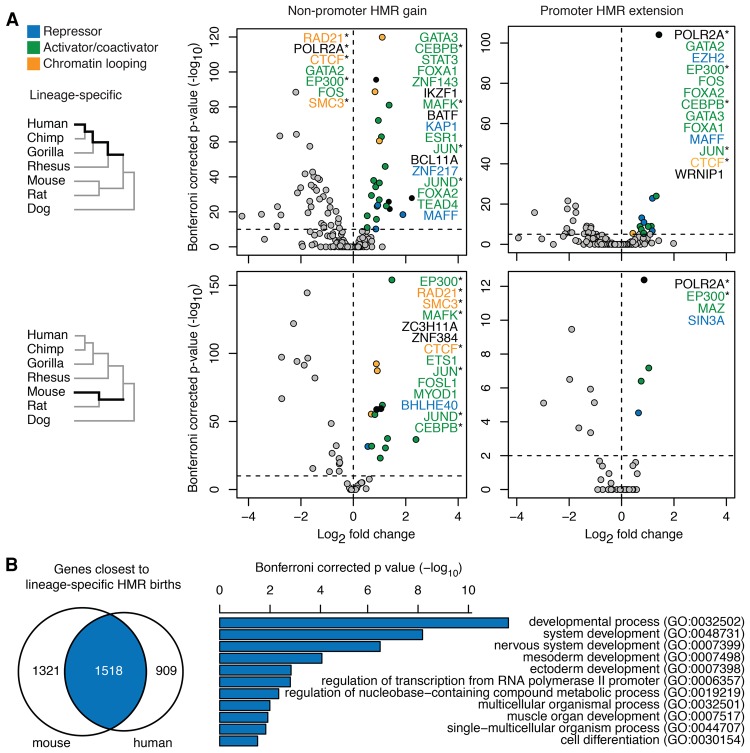
DNA methylation in the germline is among the most important factors influencing the evolution of mammalian genomes. Yet little is known about its evolutionary rate or the fraction of the methylome that has undergone change. We compared whole-genome, single-CpG DNA methylation profiles in sperm of seven species-human, chimpanzee, gorilla, rhesus macaque, mouse, rat, and dog-to investigate epigenomic evolution. We developed a phylo-epigenetic model for DNA methylation that accommodates the correlation of states at neighboring sites and allows for inference of ancestral states. Applying this model to the sperm methylomes, we uncovered an overall evolutionary expansion of the hypomethylated fraction of the genome, driven both by the birth of new hypomethylated regions and by extensive widening of hypomethylated intervals in ancestral species. This expansion shows strong lineage-specific aspects, most notably that hypomethylated intervals around transcription start sites have evolved to be considerably wider in primates and dog than in rodents, whereas rodents show evidence of a greater trend toward birth of new hypomethylated regions. Lineage-specific hypomethylated regions are enriched near sets of genes with common developmental functions and significant overlap across lineages. Rodent-specific and primate-specific hypomethylated regions are enriched for binding sites of similar transcription factors, suggesting that the plasticity accommodated by certain regulatory factors is conserved, despite substantial change in the specific sites of regulation. Overall our results reveal substantial global epigenomic change in mammalian sperm methylomes and point to a divergence in trans-epigenetic mechanisms that govern the organization of epigenetic states at gene promoters.
© 2018 Qu et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Arnosti DN, Kulkarni MM. 2005. Transcriptional enhancers: intelligent enhanceosomes or flexible billboards? J Cell Biochem 94: 890–898. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases