

Systematic screening of generic drugs for progressive multiple sclerosis identifies clomipramine as a promising therapeutic
- PMID: 29259169
- PMCID: PMC5736601
- DOI: 10.1038/s41467-017-02119-6
Systematic screening of generic drugs for progressive multiple sclerosis identifies clomipramine as a promising therapeutic
Abstract
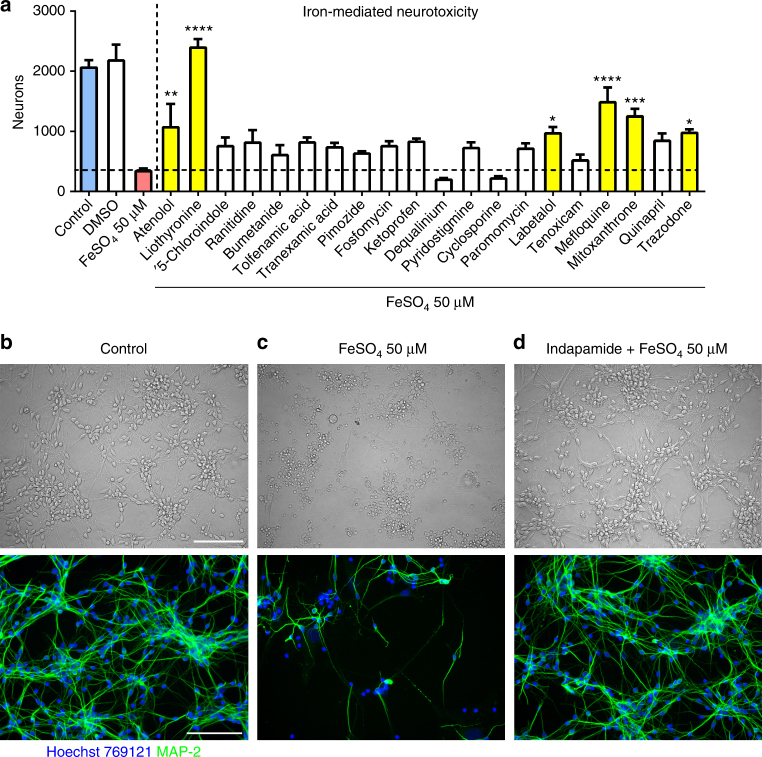
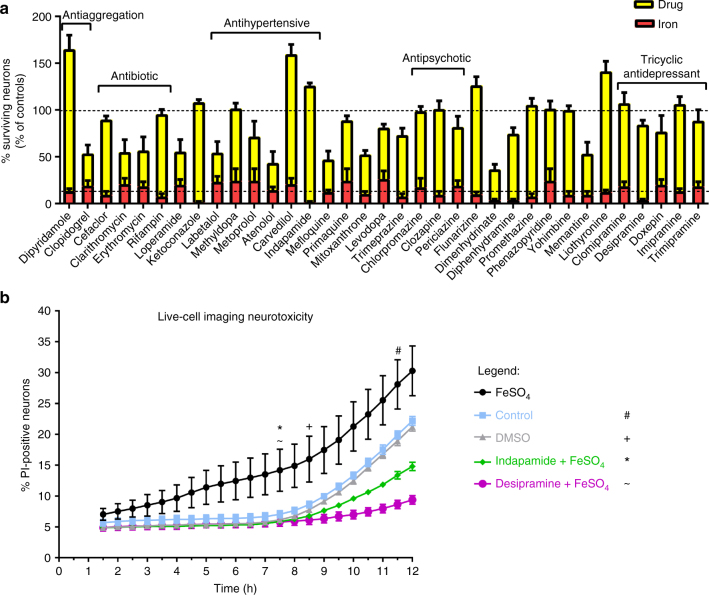
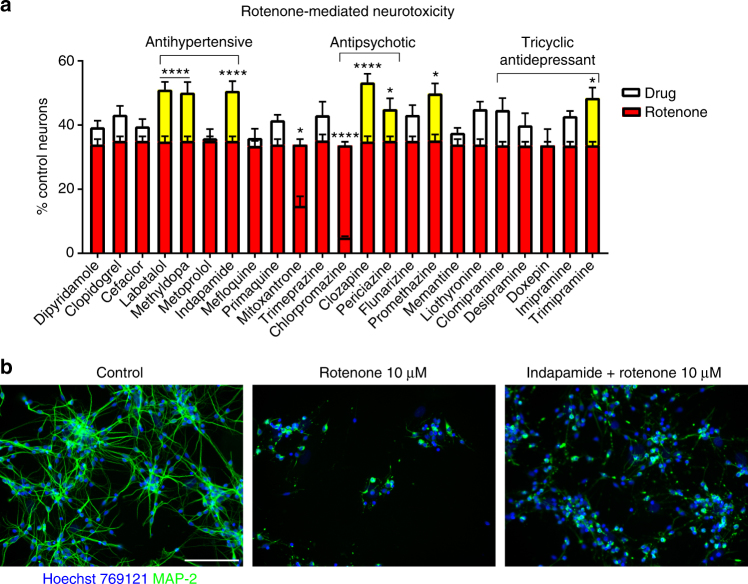
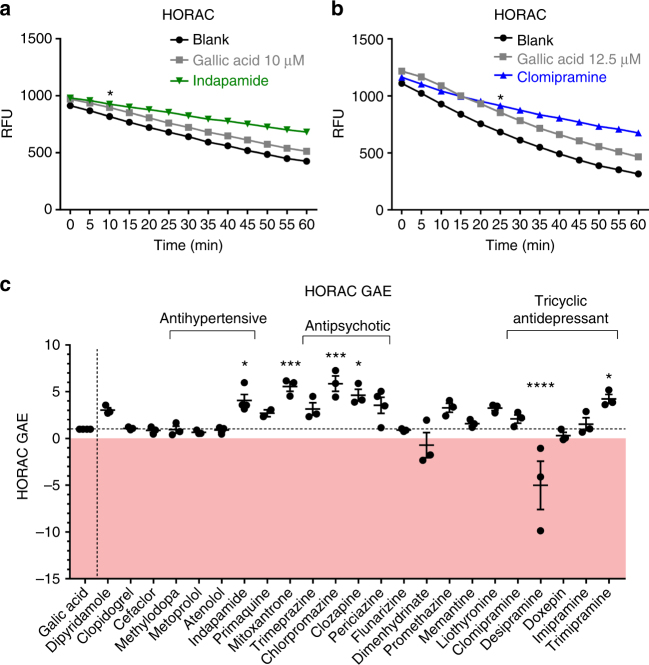
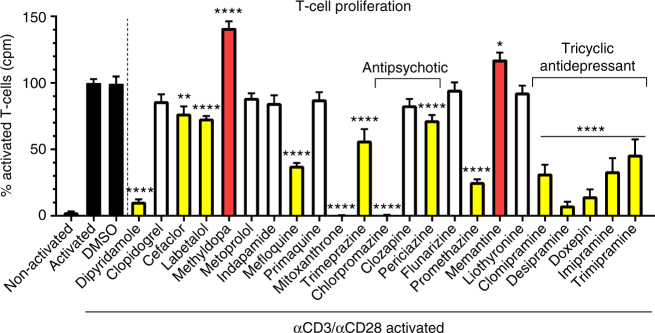
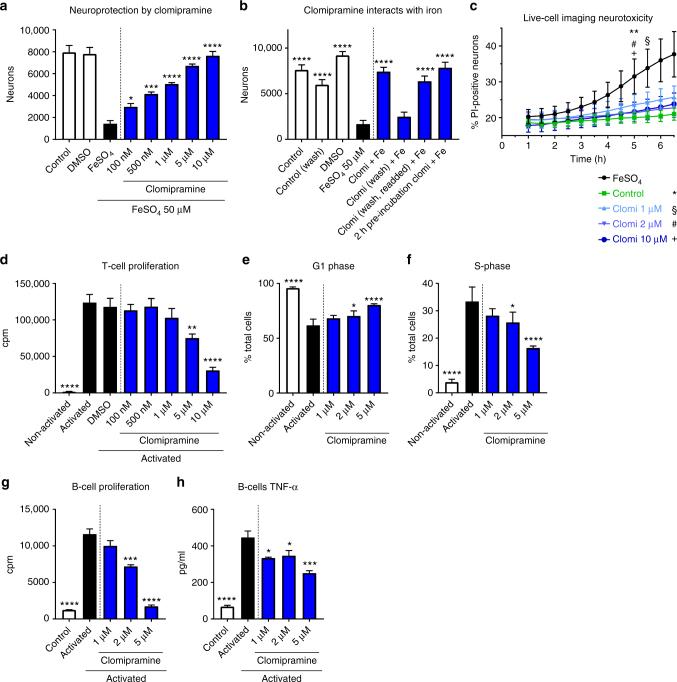
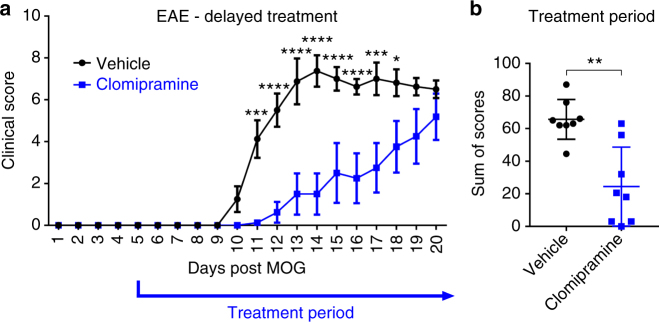
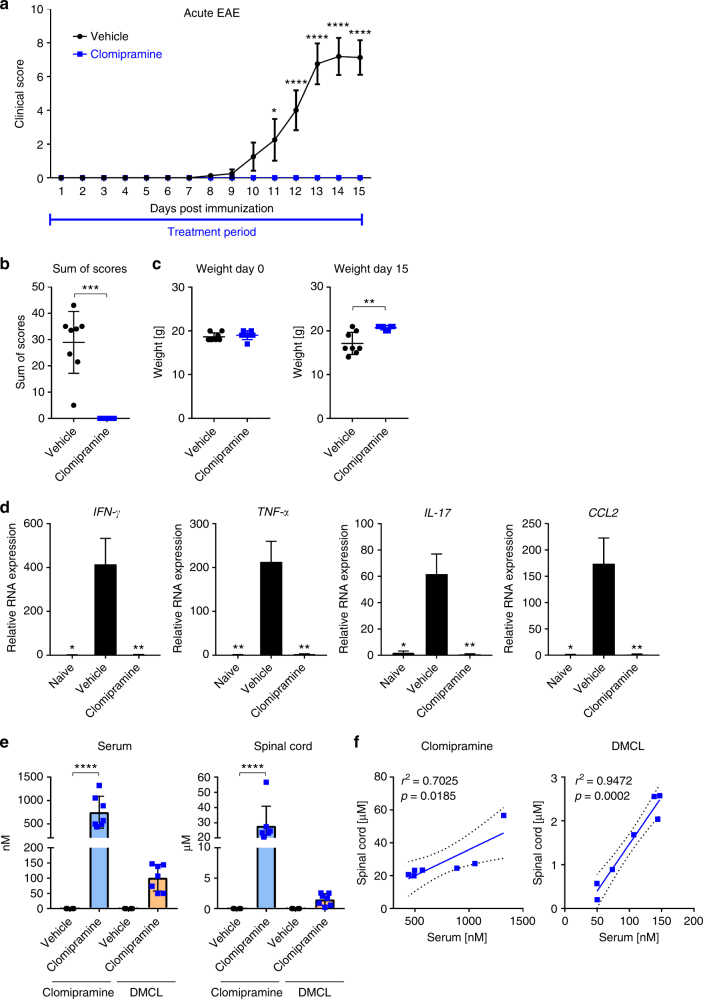
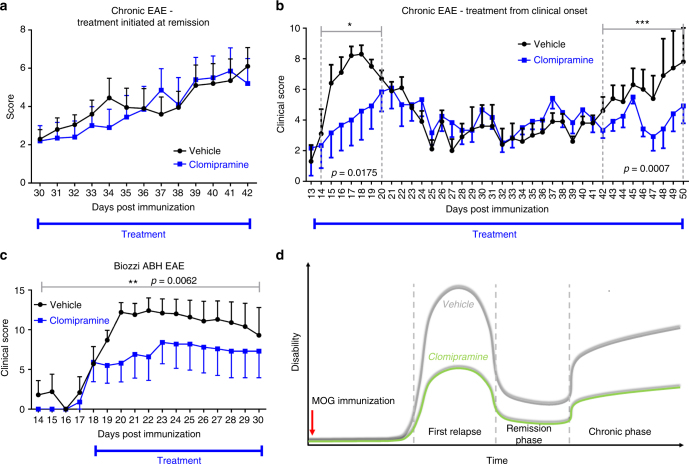
The treatment of progressive multiple sclerosis (MS) is unsatisfactory. One reason is that the drivers of disease, which include iron-mediated neurotoxicity, lymphocyte activity, and oxidative stress, are not simultaneously targeted. Here we present a systematic screen to identify generic, orally available medications that target features of progressive MS. Of 249 medications that cross the blood-brain barrier, 35 prevent iron-mediated neurotoxicity in culture. Of these, several antipsychotics and antidepressants strongly reduce T-cell proliferation and oxidative stress. We focus on the antidepressant clomipramine and found that it additionally inhibits B-lymphocyte activity. In mice with experimental autoimmune encephalomyelitis, a model of MS, clomipramine ameliorates clinical signs of acute and chronic phases. Histologically, clomipramine reduces inflammation and microglial activation, and preserves axonal integrity. In summary, we present a systematic approach to identify generic medications for progressive multiple sclerosis with the potential to advance rapidly into clinical trials, and we highlight clomipramine for further development.
Conflict of interest statement
S.F., M.K., and V.W.Y. filed a provisional patent application at the US FDA. The remaining authors declare no competing financial interests.
Figures

References
-
- Kappos L. et al. Efficacy and safety of siponimod in secondary progressive multiple sclerosis—results of the placebo controlled, double-blind, Phase III EXPAND study. ECTRIMS Online Library (2016).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
