

Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure
- PMID: 29259172
- PMCID: PMC5736591
- DOI: 10.1038/s41467-017-02292-8
Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure
Abstract
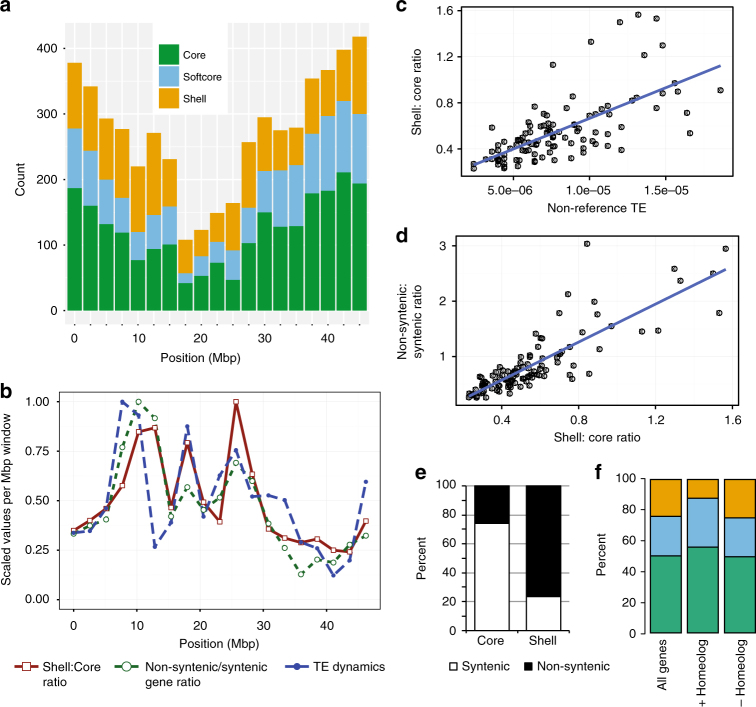
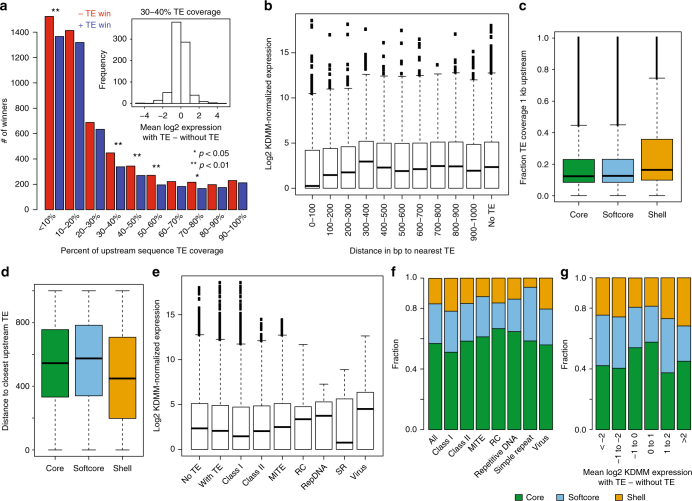
While prokaryotic pan-genomes have been shown to contain many more genes than any individual organism, the prevalence and functional significance of differentially present genes in eukaryotes remains poorly understood. Whole-genome de novo assembly and annotation of 54 lines of the grass Brachypodium distachyon yield a pan-genome containing nearly twice the number of genes found in any individual genome. Genes present in all lines are enriched for essential biological functions, while genes present in only some lines are enriched for conditionally beneficial functions (e.g., defense and development), display faster evolutionary rates, lie closer to transposable elements and are less likely to be syntenic with orthologous genes in other grasses. Our data suggest that differentially present genes contribute substantially to phenotypic variation within a eukaryote species, these genes have a major influence in population genetics, and transposable elements play a key role in pan-genome evolution.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
