

Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974-2015
- PMID: 29259596
- PMCID: PMC5723339
- DOI: 10.3389/fmicb.2017.02399
Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974-2015
Abstract
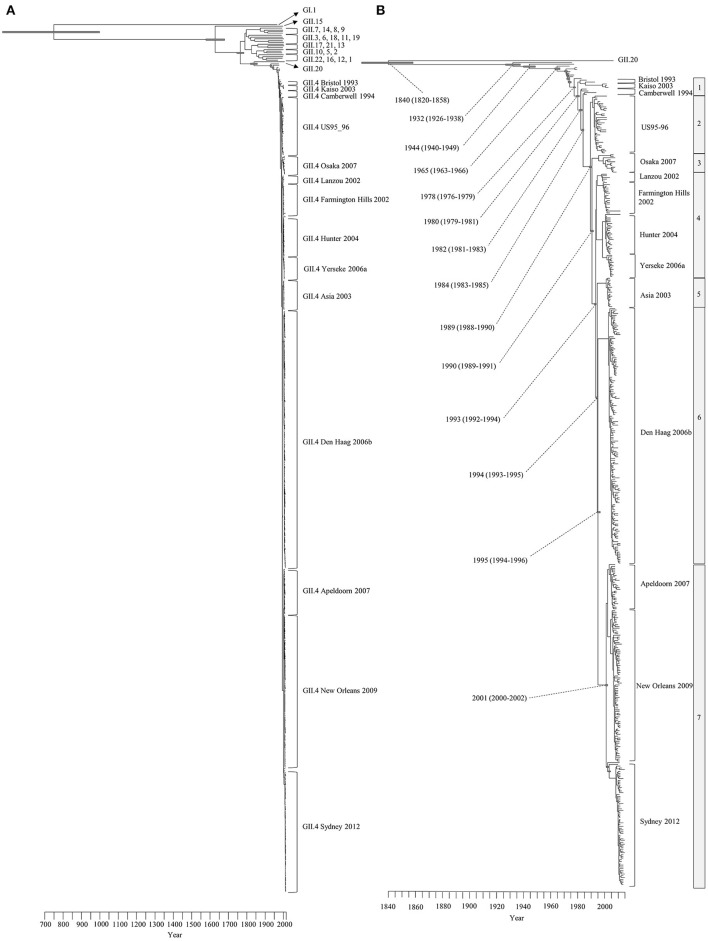
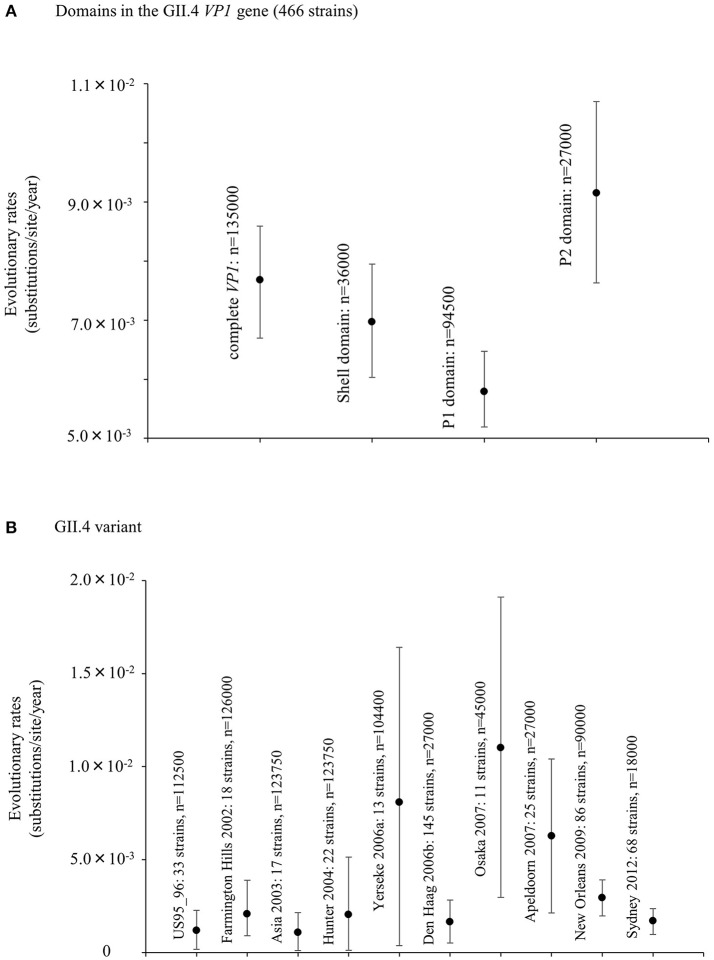
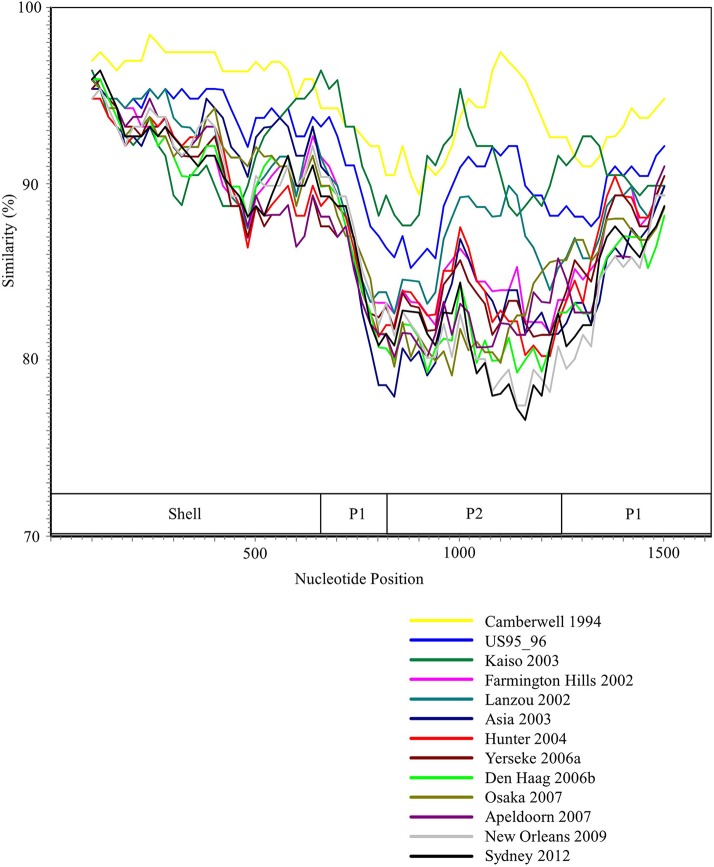
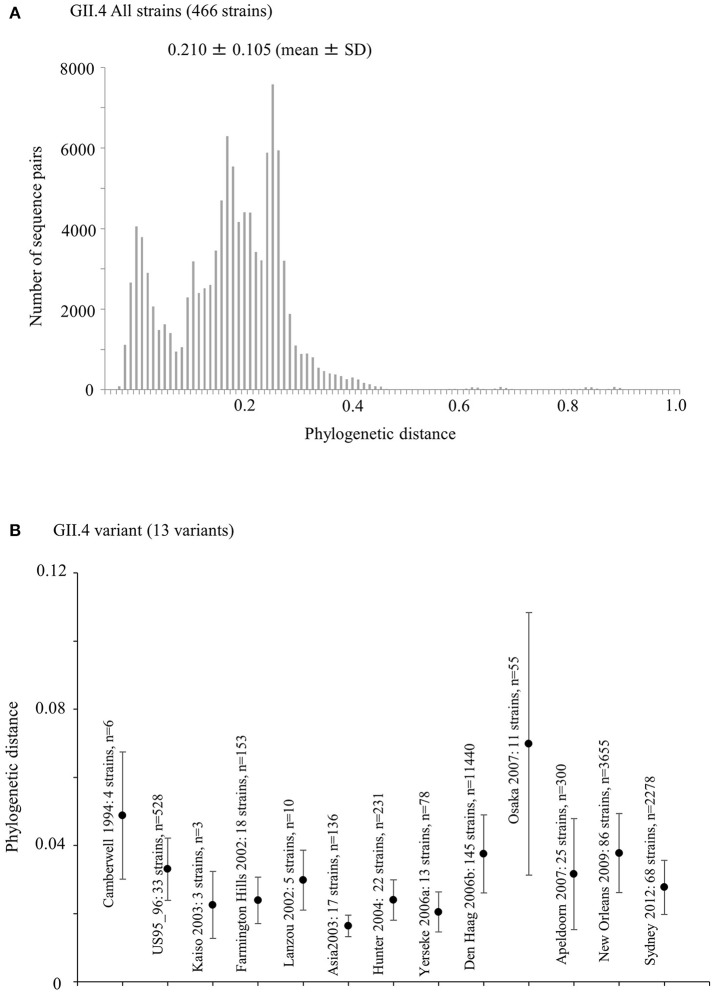
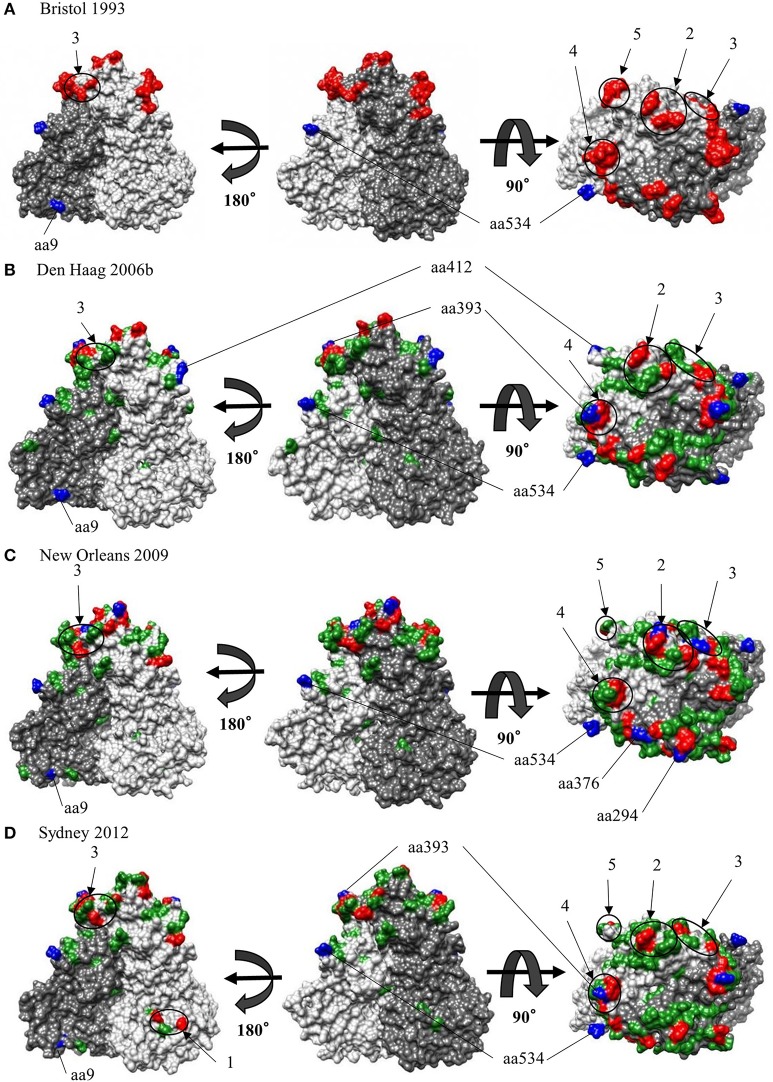
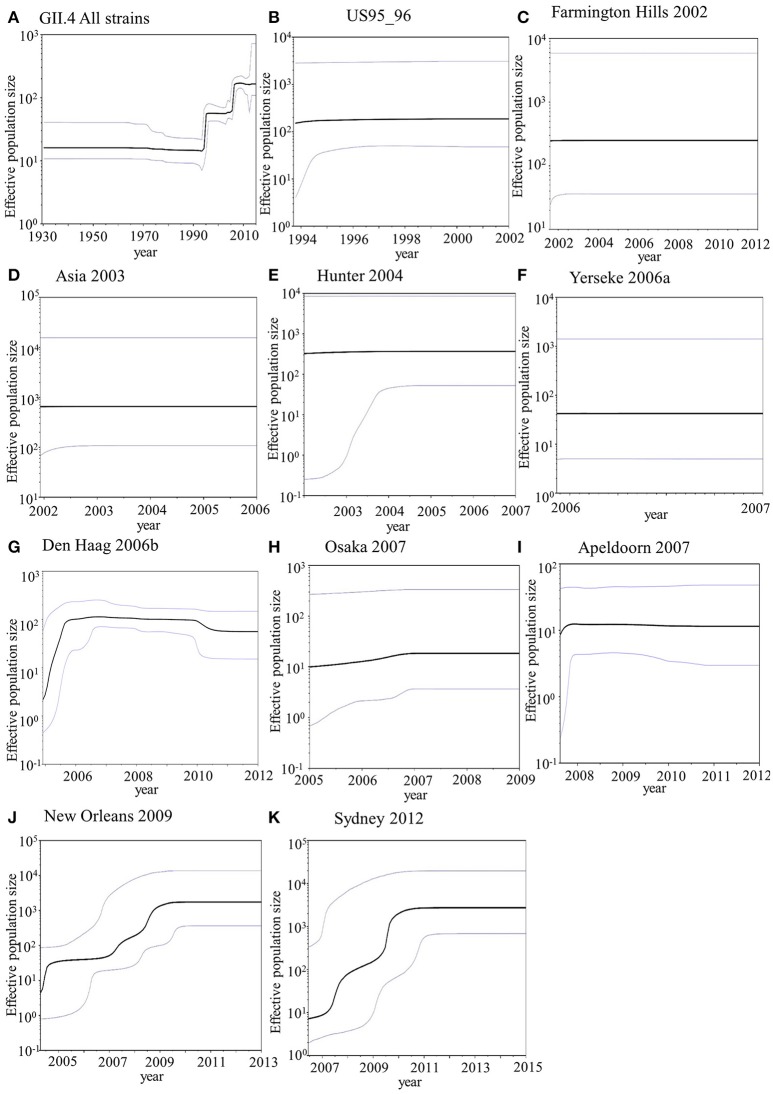
Human norovirus (HuNoV) is a leading cause of viral gastroenteritis worldwide, of which GII.4 is the most predominant genotype. Unlike other genotypes, GII.4 has created various variants that escaped from previously acquired immunity of the host and caused repeated epidemics. However, the molecular evolutionary differences among all GII.4 variants, including recently discovered strains, have not been elucidated. Thus, we conducted a series of bioinformatic analyses using numerous, globally collected, full-length GII.4 major capsid (VP1) gene sequences (466 strains) to compare the evolutionary patterns among GII.4 variants. The time-scaled phylogenetic tree constructed using the Bayesian Markov chain Monte Carlo (MCMC) method showed that the common ancestor of the GII.4 VP1 gene diverged from GII.20 in 1840. The GII.4 genotype emerged in 1932, and then formed seven clusters including 14 known variants after 1980. The evolutionary rate of GII.4 strains was estimated to be 7.68 × 10-3 substitutions/site/year. The evolutionary rates probably differed among variants as well as domains [protruding 1 (P1), shell, and P2 domains]. The Osaka 2007 variant strains probably contained more nucleotide substitutions than any other variant. Few conformational epitopes were located in the shell and P1 domains, although most were contained in the P2 domain, which, as previously established, is associated with attachment to host factors and antigenicity. We found that positive selection sites for the whole GII.4 genotype existed in the shell and P1 domains, while Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants were under positive selection in the P2 domain. Amino acid substitutions overlapped with putative epitopes or were located around the epitopes in the P2 domain. The effective population sizes of the present strains increased stepwise for Den Haag 2006b, New Orleans 2009, and Sydney 2012 variants. These results suggest that HuNoV GII.4 rapidly evolved in a few decades, created various variants, and altered its evolutionary rate and antigenicity.
Keywords: GII.4; Norovirus; VP1; bioinformatics; molecular evolution.
Figures
References
-
- Añez G., Grinev A., Chancey C., Ball C., Akolkar N., Land K. J., et al. (2013). Evolutionary dynamics of West Nile virus in the United States, 1999-2011: phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 7:e2245. 10.1371/journal.pntd.0002245 - DOI - PMC - PubMed
-
- Allen D. J., Trainor E., Callaghan A., O'Brien S. J., Cunliffe N. A., Iturriza-Gómara M. (2016). Early detection of epidemic GII-4 norovirus strains in UK and Malawi: role of surveillance of sporadic acute gastroenteritis in anticipating global epidemics. PLoS ONE 11:e0146972. 10.1371/journal.pone.0146972 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources