

RB1 gene mutations in Argentine retinoblastoma patients. Implications for genetic counseling
- PMID: 29261756
- PMCID: PMC5738096
- DOI: 10.1371/journal.pone.0189736
RB1 gene mutations in Argentine retinoblastoma patients. Implications for genetic counseling
Abstract
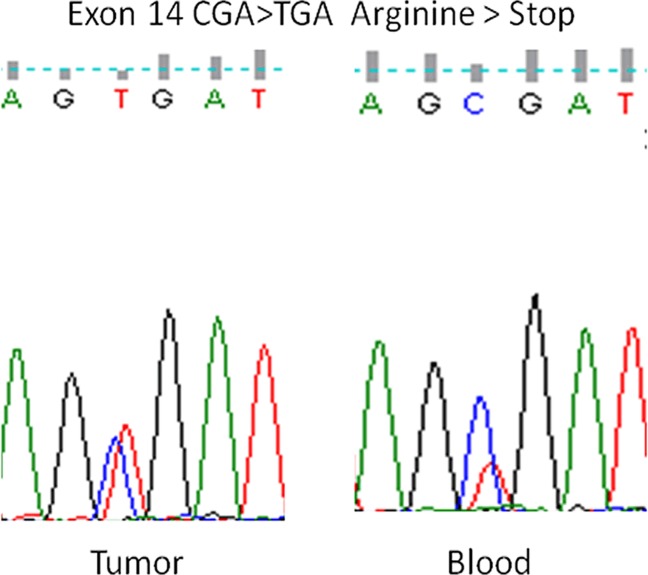
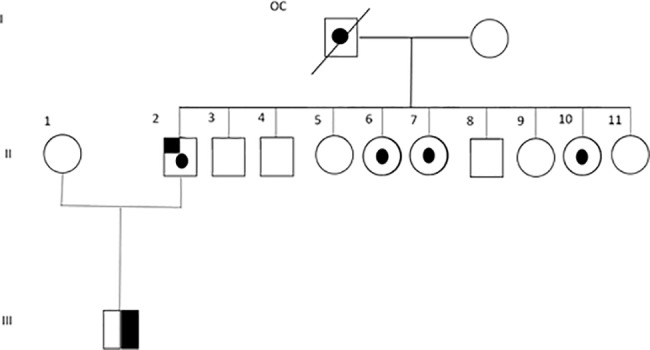
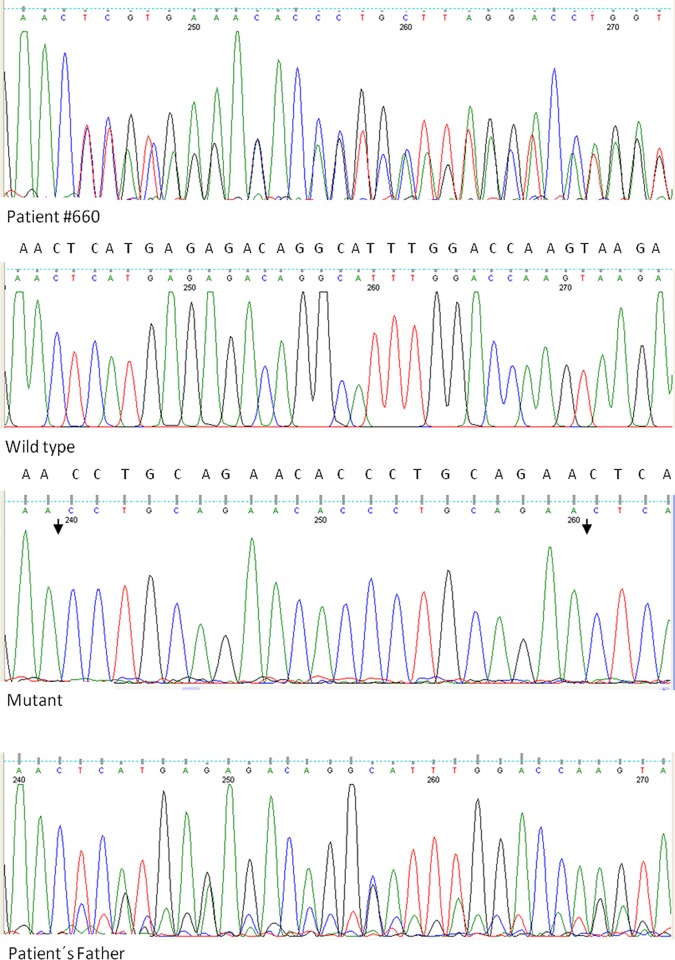
Retinoblastoma (RB) is an inherited childhood ocular cancer caused by mutations in the tumor suppressor RB1 gene. Identification of RB1 mutations is essential to assess the risk of developing retinoblastoma in the patients´ relatives. Retinoblastoma is a potentially curable cancer and an early diagnosis is critical for survival and eye preservation. Unilateral retinoblastoma is mostly non-heritable and results from two somatic mutations whereas bilateral retinoblastoma is heritable and results from one germline and one somatic mutation, both have high penetrance, 90%. The purpose of this study was to identify causative RB1 mutations in RB patients with different clinical presentations. A comprehensive approach was used to study a cohort of 34 patients with unilateral, bilateral and trilateral retinoblastoma. Blood and tumor DNA was analyzed by sequencing and multiplex ligation-dependent probe amplification (MLPA) assay. Validation of an insertion mutation was performed by cloning the PCR product. Most of the patients in our cohort had unilateral RB, eight patients had bilateral RB and one patient had a trilateral tumor with ocular and suprasellar/sellar locations. Other tumors in addition to retinoblastoma were also found in the affected families. One patient had two syndromes, retinoblastoma and schwannomatosis, and another RB patient had a father with a retinoma. Five out of the 25 unilateral RB patients carried germinal mutations (20%), which were mostly missense mutations. The bilateral and trilateral patients carried splice-site, nonsense and frameshift mutations as well as a whole RB1 gene deletion. Missense mutations were associated with mild phenotype: unilateral retinoblastoma, retinoma or no tumor. In this study we identified causative RB1 mutations in most bilateral RB patients and in some unilateral RB patients, including five novel mutations. These data are crucial for genetic counseling and confirm the need to perform complete genetic screening for RB1 mutations in both constitutional and tumor tissues.
Conflict of interest statement
Figures
References
-
- Gallie BL, Campbell C, Devlin H, Duckett A, Squire JA Developmental basis of retinal-specific induction of cancer by RB mutation.Cancer Res 1999; 1[Suppl 7]:1731s–1735s. - PubMed
-
- Abramson DH, Beaverson K, Sangani P, Vora RA, Lee TC, Hochberg HM et al. Screening for retinoblastoma: presentingsigns as prognosticators of patient and ocular survival. Pediatrics 2003; 112: 1248–1255. - PubMed
-
- Vogel F. Genetics of retinoblastoma. Hum Genet; 1979; 52:1–54. - PubMed
-
- Chantada GL, Qaddoumi I, Canturk S, Ketan Vikas, Zhigui MA, Kimani Kahaki et al. Strategies to manage retinoblastoma in developing countries. Pediatr Blood Cancer. 2011;56:341–348. doi: 10.1002/pbc.22843 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
