

A homozygous mutation in the stem II domain of RNU4ATAC causes typical Roifman syndrome
- PMID: 29263834
- PMCID: PMC5677950
- DOI: 10.1038/s41525-017-0024-5
A homozygous mutation in the stem II domain of RNU4ATAC causes typical Roifman syndrome
Abstract
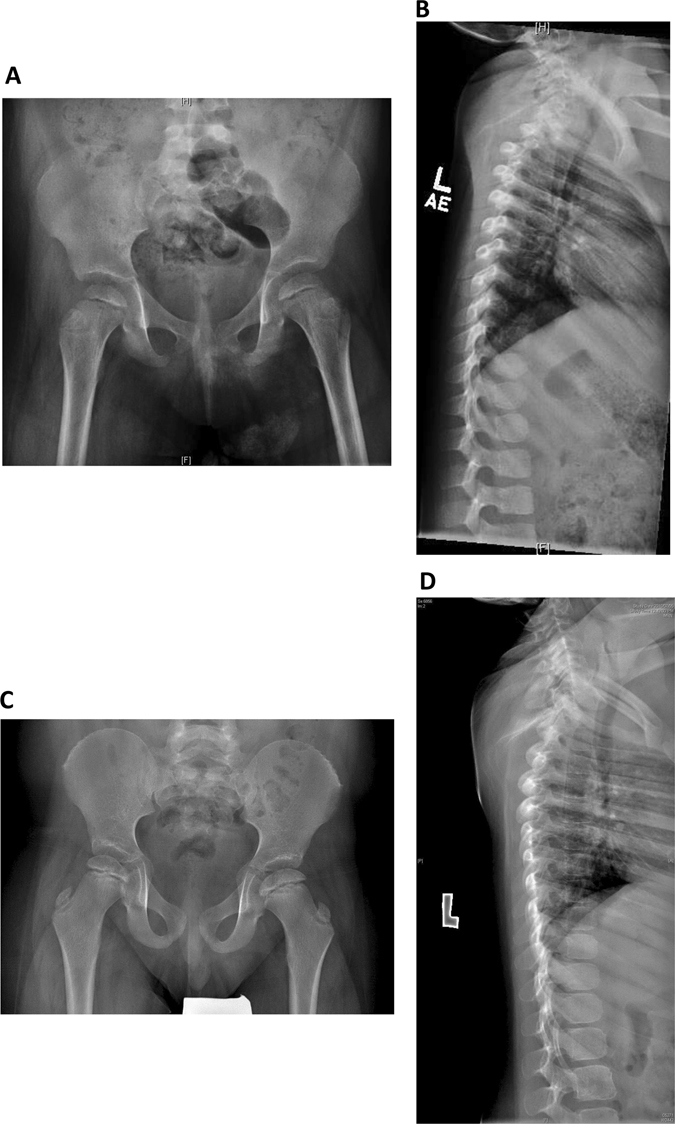
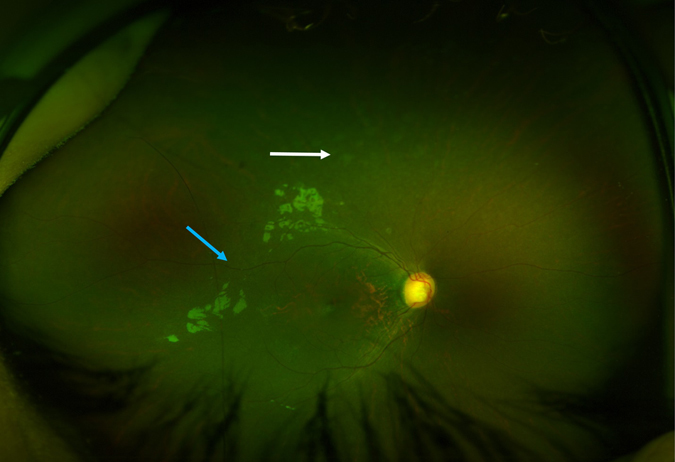
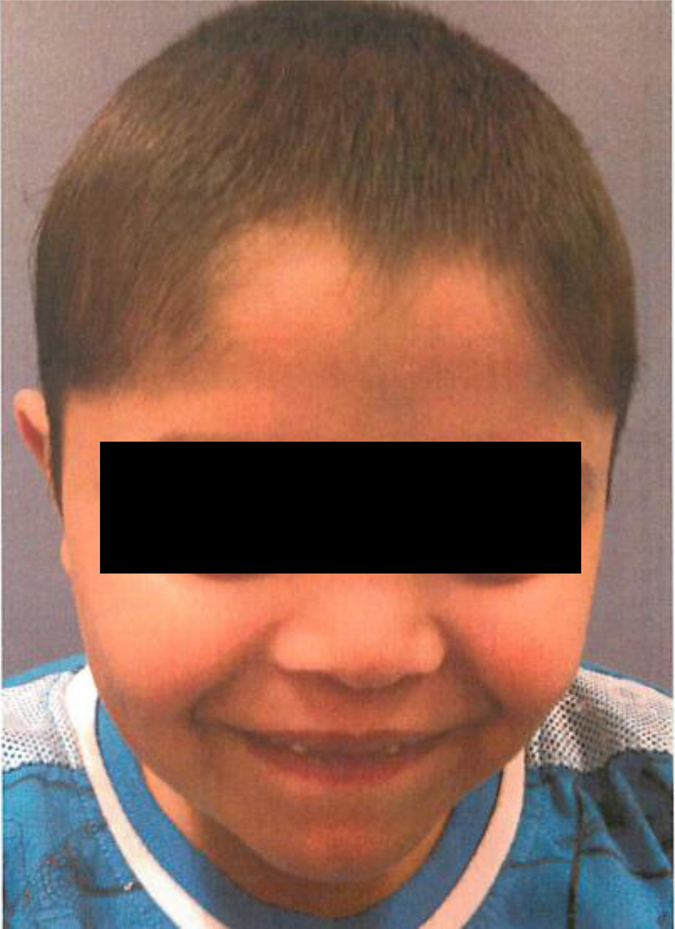
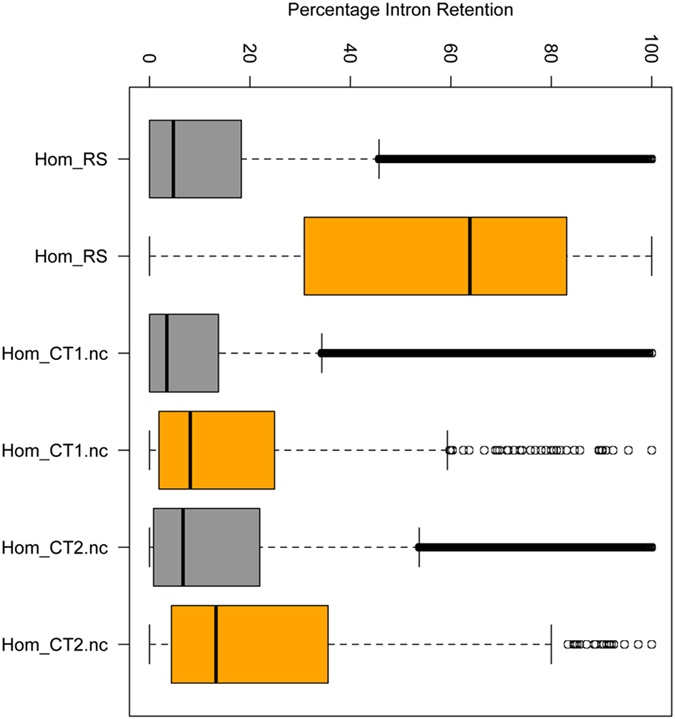
Roifman syndrome (OMIM# 616651) is a complex syndrome encompassing skeletal dysplasia, immunodeficiency, retinal dystrophy and developmental delay, and is caused by compound heterozygous mutations involving the Stem II region and one of the other domains of the RNU4ATAC gene. This small nuclear RNA gene is essential for minor intron splicing. The Canadian Centre for Primary Immunodeficiency Registry and Repository were used to derive patient information as well as tissues. Utilising RNA sequencing methodologies, we analysed samples from patients with Roifman syndrome and assessed intron retention. We demonstrate that a homozygous mutation in Stem II is sufficient to cause the full spectrum of features associated with typical Roifman syndrome. Further, we demonstrate the same pattern of aberration in minor intron retention as found in cases with compound heterozygous mutations.
Conflict of interest statement
The authors declare that they have no competing financial interests.Medical records were compiled into the Canadian Centre for Primary Immunodeficiency Registry, which has been approved by the Sick Kids Research Ethics board (protocol number 1000005598). This includes consent and assent from patients and family members for genetics analysis.
Figures


References
-
- Roifman CM. Immunological aspects of a novel immunodeficiency syndrome that includes antibody deficiency with normal immunoglobulins, spondyloepiphyseal dysplasia, growth and developmental delay, and retinal dystrophy. Can. J. Allergy Clini. Immunol. 1997;2:94–98.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
