

A framework to identify contributing genes in patients with Phelan-McDermid syndrome
- PMID: 29263841
- PMCID: PMC5677962
- DOI: 10.1038/s41525-017-0035-2
A framework to identify contributing genes in patients with Phelan-McDermid syndrome
Erratum in
-
Erratum: Author Correction: A framework to identify contributing genes in patients with Phelan-McDermid syndrome.NPJ Genom Med. 2019 Jul 1;4:16. doi: 10.1038/s41525-019-0090-y. eCollection 2019. NPJ Genom Med. 2019. PMID: 31285849 Free PMC article.
Abstract
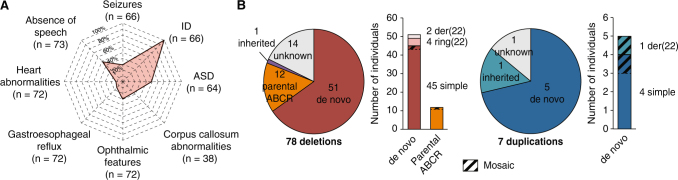
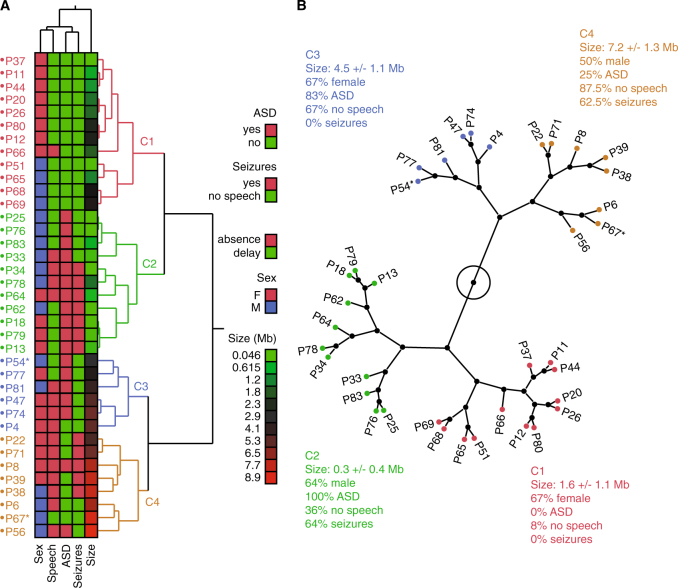
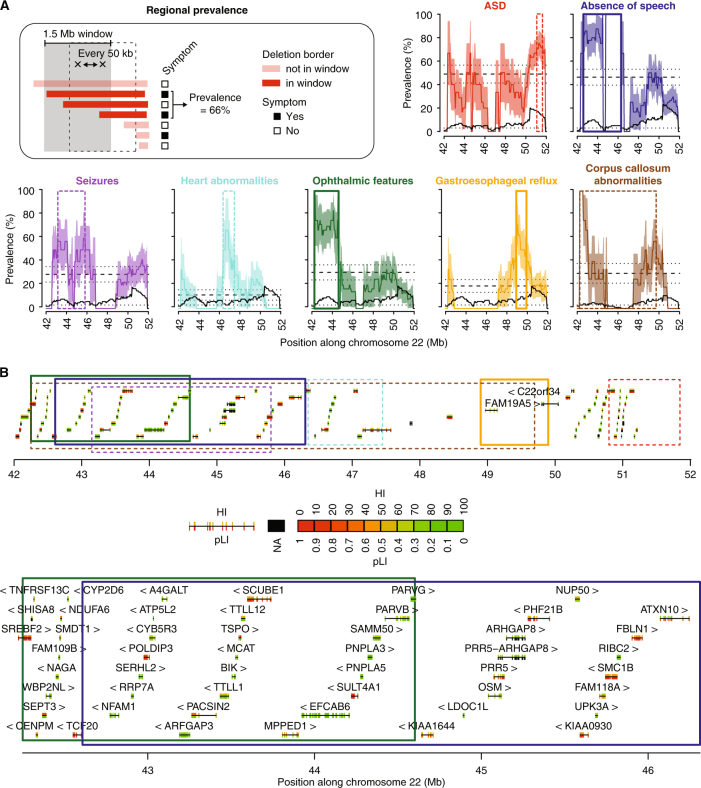
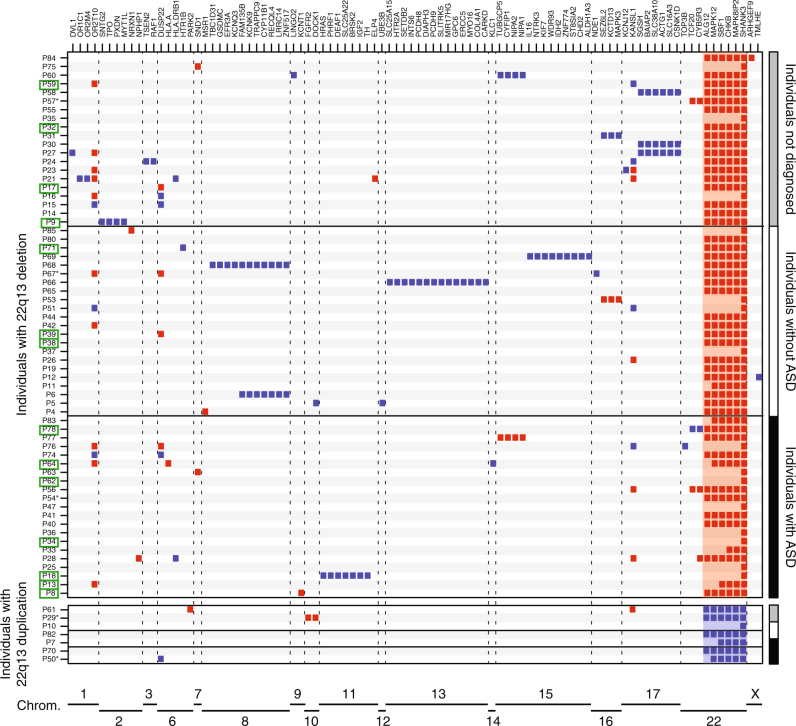
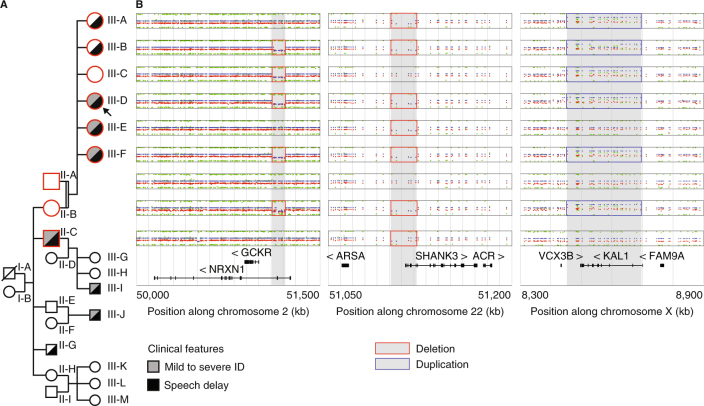
Phelan-McDermid syndrome (PMS) is characterized by a variety of clinical symptoms with heterogeneous degrees of severity, including intellectual disability (ID), absent or delayed speech, and autism spectrum disorders (ASD). It results from a deletion of the distal part of chromosome 22q13 that in most cases includes the SHANK3 gene. SHANK3 is considered a major gene for PMS, but the factors that modulate the severity of the syndrome remain largely unknown. In this study, we investigated 85 patients with different 22q13 rearrangements (78 deletions and 7 duplications). We first explored the clinical features associated with PMS, and provide evidence for frequent corpus callosum abnormalities in 28% of 35 patients with brain imaging data. We then mapped several candidate genomic regions at the 22q13 region associated with high risk of clinical features, and suggest a second locus at 22q13 associated with absence of speech. Finally, in some cases, we identified additional clinically relevant copy-number variants (CNVs) at loci associated with ASD, such as 16p11.2 and 15q11q13, which could modulate the severity of the syndrome. We also report an inherited SHANK3 deletion transmitted to five affected daughters by a mother without ID nor ASD, suggesting that some individuals could compensate for such mutations. In summary, we shed light on the genotype-phenotype relationship of patients with PMS, a step towards the identification of compensatory mechanisms for a better prognosis and possibly treatments of patients with neurodevelopmental disorders.
Conflict of interest statement
The authors declare that they have no competing financial interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
