

Germline and somatic mutations in STXBP1 with diverse neurodevelopmental phenotypes
- PMID: 29264391
- PMCID: PMC5735305
- DOI: 10.1212/NXG.0000000000000199
Germline and somatic mutations in STXBP1 with diverse neurodevelopmental phenotypes
Abstract
Objective: To expand the clinical phenotype associated with STXBP1 gene mutations and to understand the effect of STXBP1 mutations in the pathogenesis of focal cortical dysplasia (FCD).
Methods: Patients with STXBP1 mutations were identified in various ways: as part of a retrospective cohort study of epileptic encephalopathy; through clinical referrals of individuals (10,619) with developmental delay (DD) for chromosomal microarray; and from a collection of 5,205 individuals with autism spectrum disorder (ASD) examined by whole-genome sequencing.
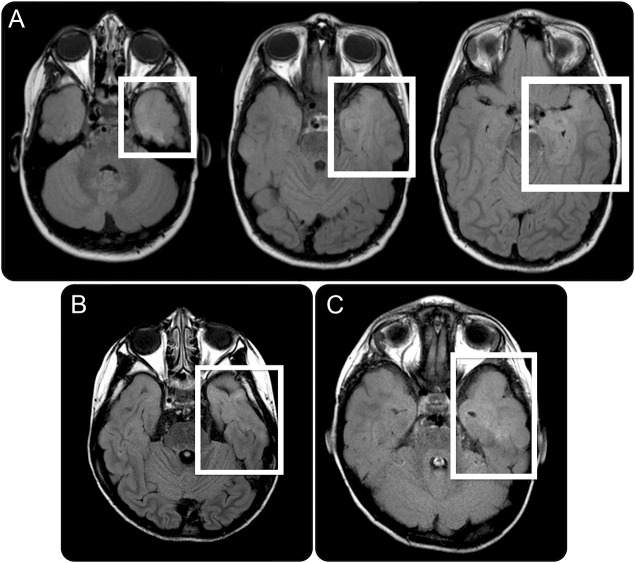
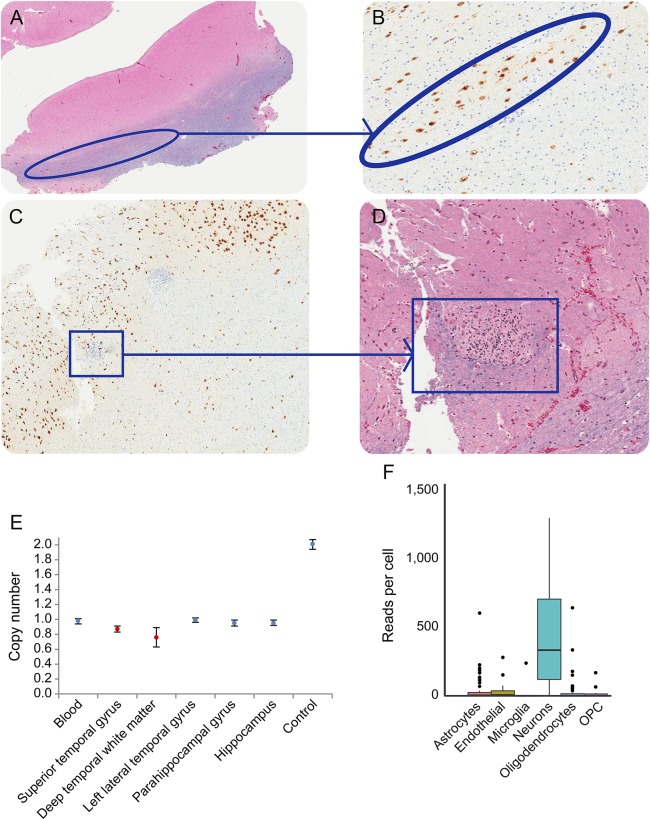
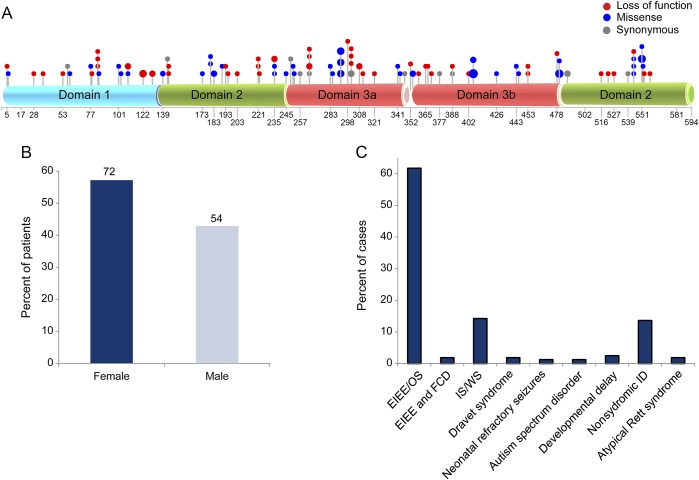
Results: Seven patients with heterozygous de novo mutations affecting the coding region of STXBP1 were newly identified. Three cases had radiologic evidence suggestive of FCD. One male patient with early infantile epileptic encephalopathy, DD, and ASD achieved complete seizure remission following resection of dysplastic brain tissue. Examination of excised brain tissue identified mosaicism for STXBP1, providing evidence for a somatic mechanism. Cell-type expression analysis suggested neuron-specific expression. A comprehensive analysis of the published data revealed that 3.1% of severe epilepsy cases carry a pathogenic de novo mutation within STXBP1. By contrast, ASD was rarely associated with mutations in this gene in our large cohorts.
Conclusions: STXBP1 mutations are an important cause of epilepsy and are also rarely associated with ASD. In a case with histologically proven FCD, an STXBP1 somatic mutation was identified, suggesting a role in its etiology. Removing such tissue may be curative for STXBP1-related epilepsy.
Figures
References
-
- Gburek-Augustat J, Beck-Woedl S, Tzschach A, Bauer P, Schoening M, Riess A. Epilepsy is not a mandatory feature of STXBP1 associated ataxia-tremor-retardation syndrome. Eur J Paediatr Neurol 2016;20:661–665. - PubMed
-
- Hamdan FF, Piton A, Gauthier J, et al. . De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009;65:748–753. - PubMed
-
- Mignot C, Moutard ML, Trouillard O, et al. . STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011;52:1820–1827. - PubMed
-
- Stamberger H, Nikanorova M, Willemsen MH, et al. . STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 2016;86:954–962. - PubMed
-
- Otsuka M, Oguni H, Liang JS, et al. . STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome–result of Japanese cohort study. Epilepsia 2010;51:2449–2452. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources