

Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism
- PMID: 29272116
- PMCID: PMC5831524
- DOI: 10.1021/acs.chemrev.7b00510
Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism
Erratum in
-
Correction to: Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism.Chem Rev. 2018 May 23;118(10):5261-5263. doi: 10.1021/acs.chemrev.8b00181. Epub 2018 Apr 5. Chem Rev. 2018. PMID: 29620360 No abstract available.
Abstract
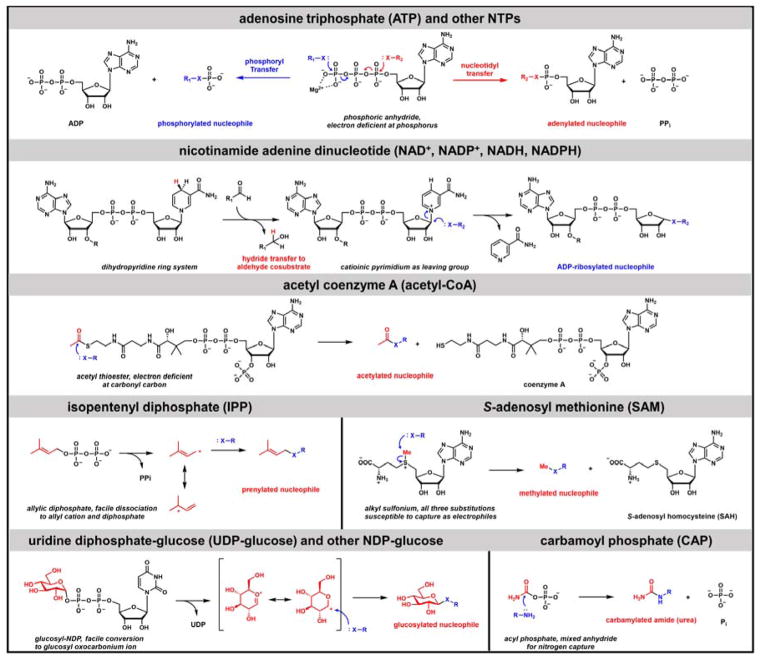
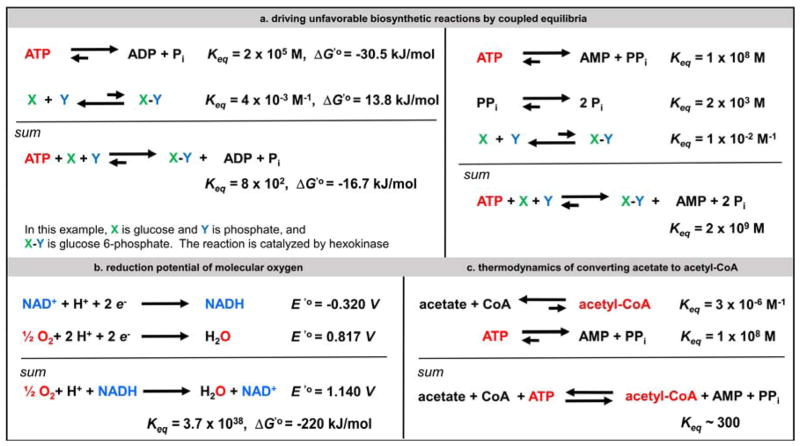
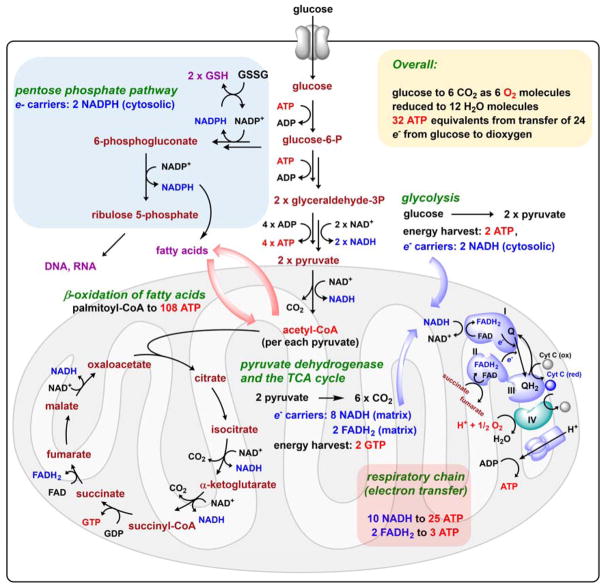
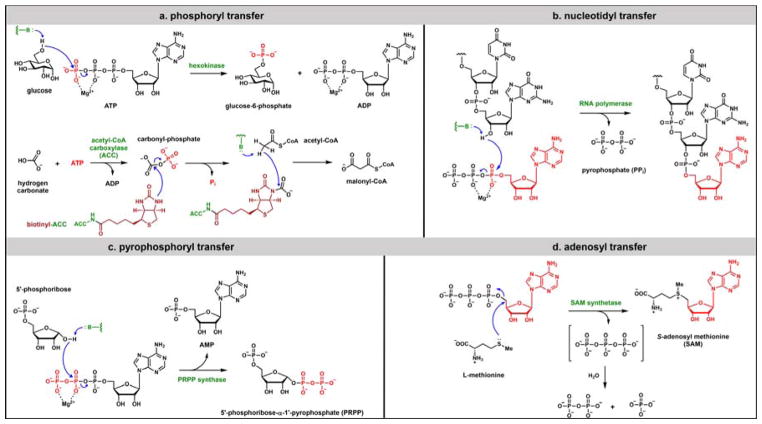
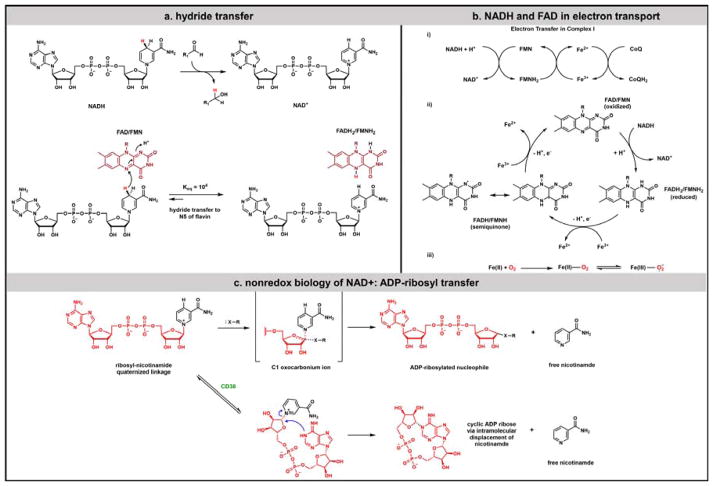
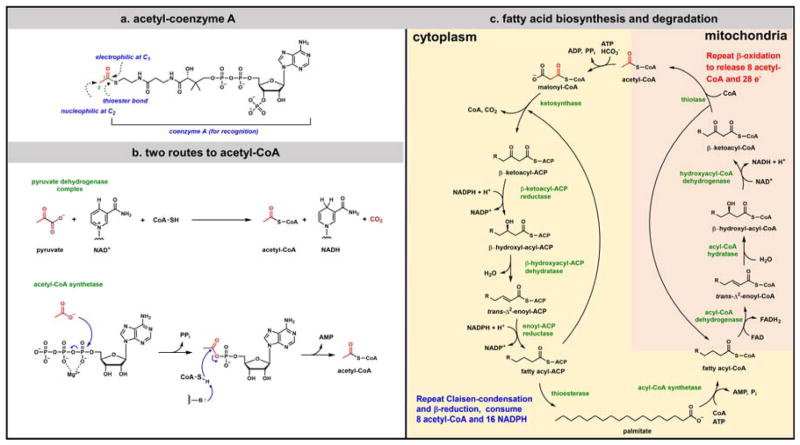
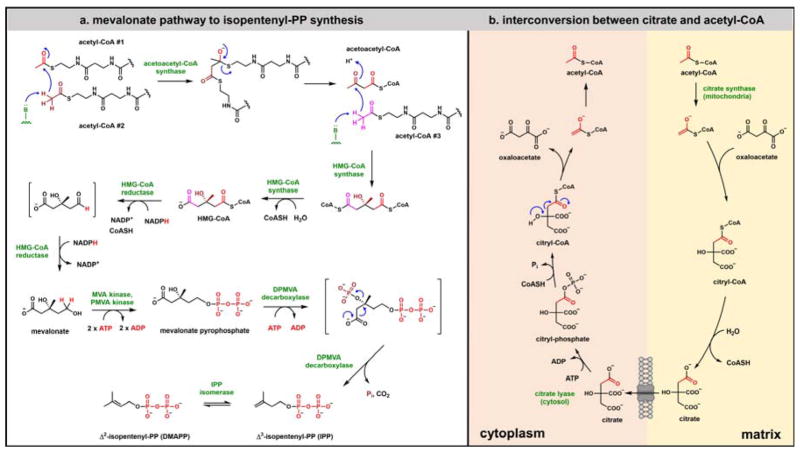
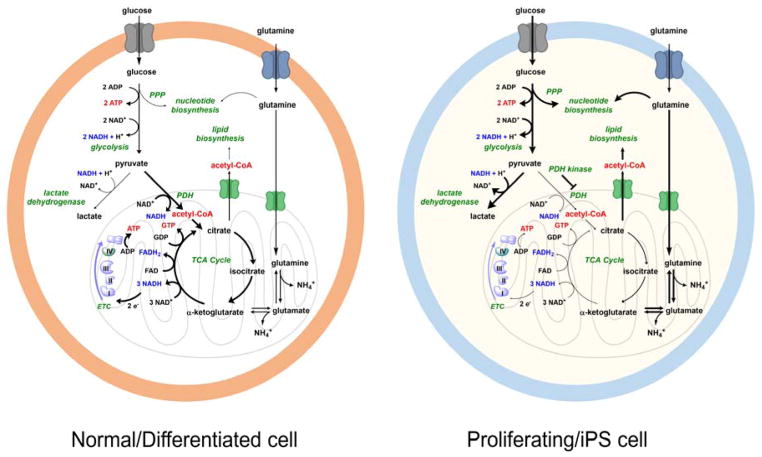
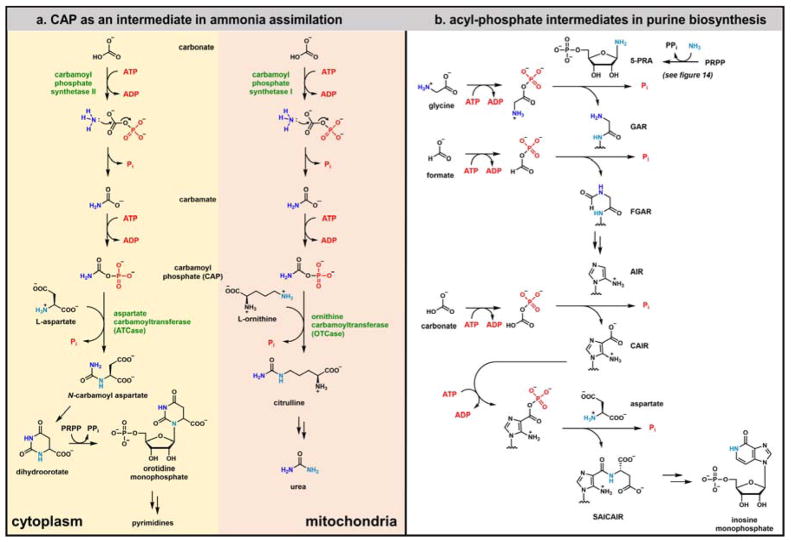
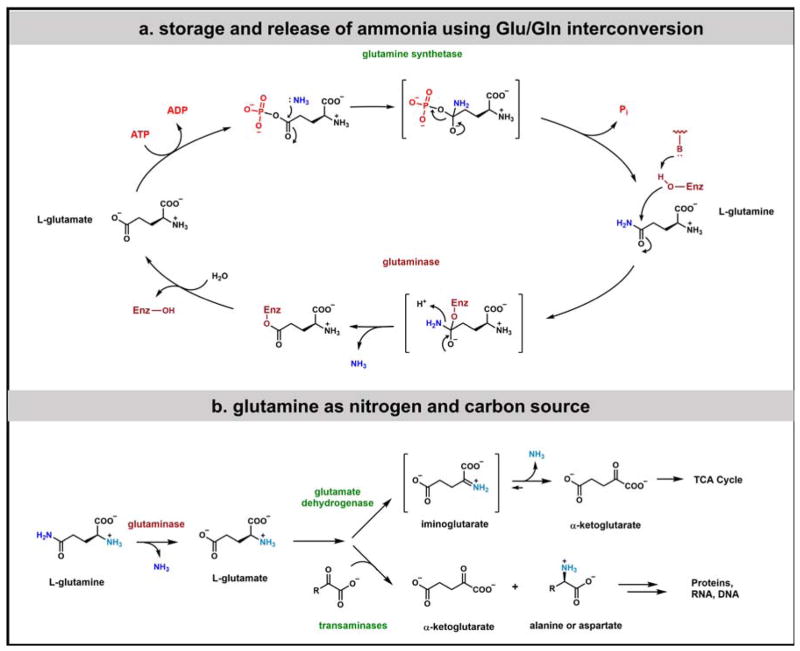
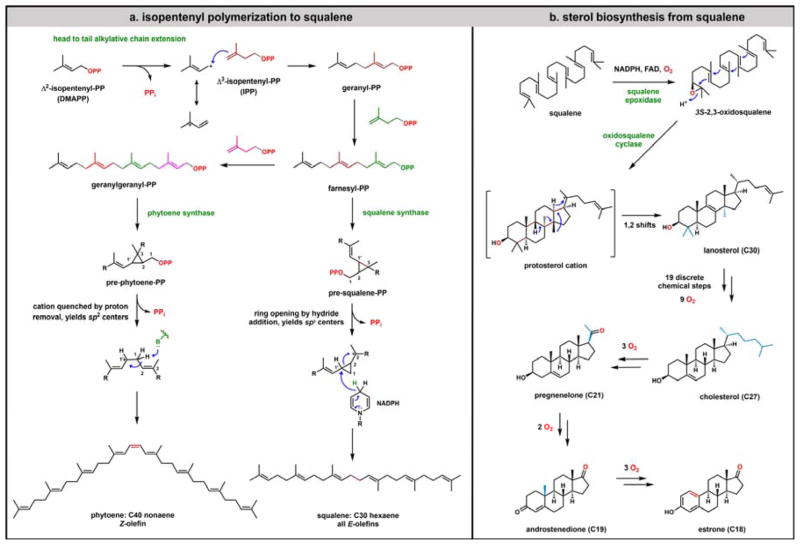
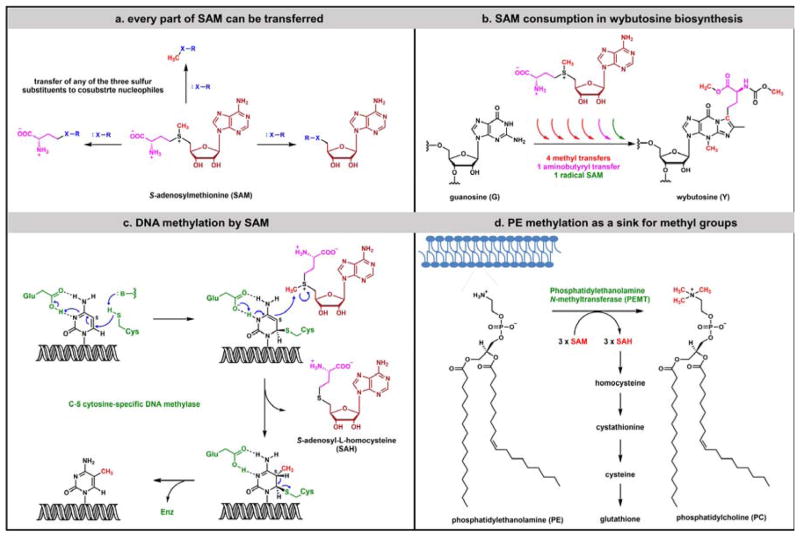
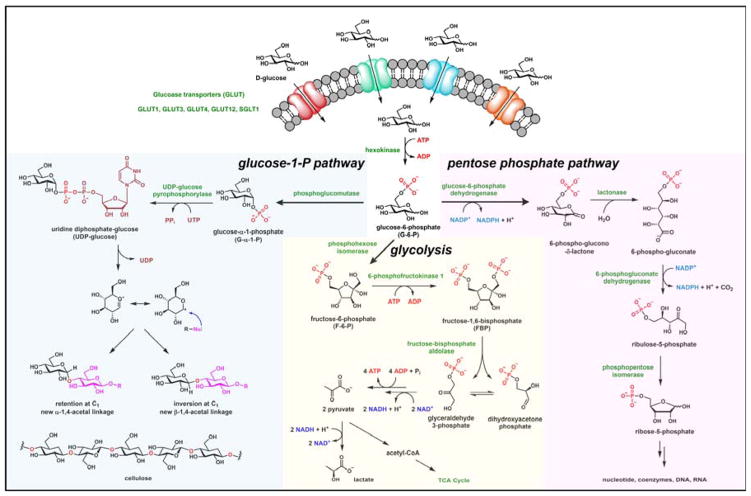
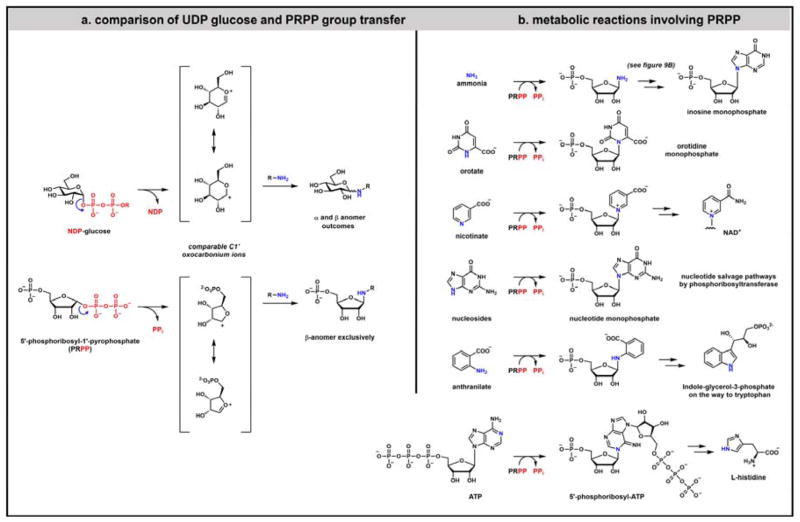
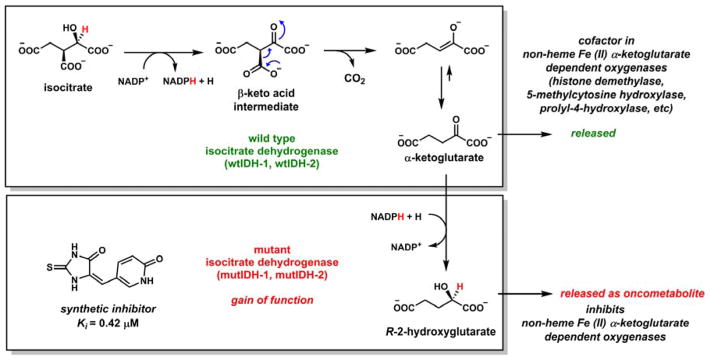
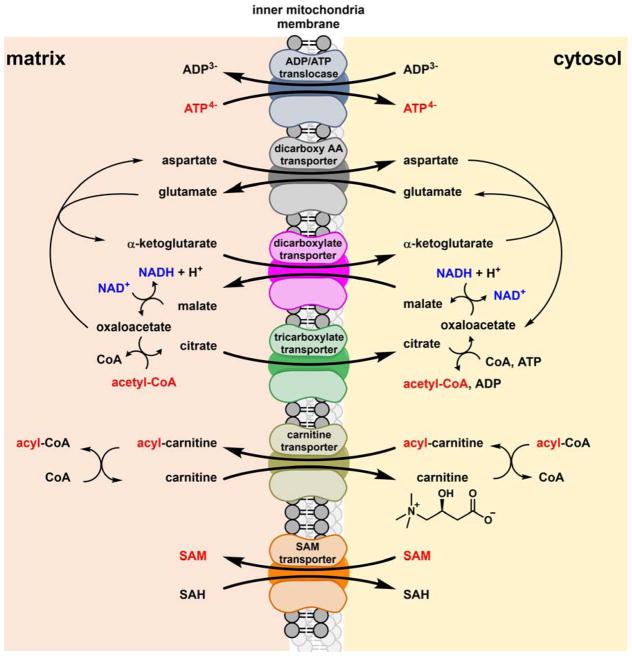
Contemporary analyses of cell metabolism have called out three metabolites: ATP, NADH, and acetyl-CoA, as sentinel molecules whose accumulation represent much of the purpose of the catabolic arms of metabolism and then drive many anabolic pathways. Such analyses largely leave out how and why ATP, NADH, and acetyl-CoA (Figure 1 ) at the molecular level play such central roles. Yet, without those insights into why cells accumulate them and how the enabling properties of these key metabolites power much of cell metabolism, the underlying molecular logic remains mysterious. Four other metabolites, S-adenosylmethionine, carbamoyl phosphate, UDP-glucose, and Δ2-isopentenyl-PP play similar roles in using group transfer chemistry to drive otherwise unfavorable biosynthetic equilibria. This review provides the underlying chemical logic to remind how these seven key molecules function as mobile packets of cellular currencies for phosphoryl transfers (ATP), acyl transfers (acetyl-CoA, carbamoyl-P), methyl transfers (SAM), prenyl transfers (IPP), glucosyl transfers (UDP-glucose), and electron and ADP-ribosyl transfers (NAD(P)H/NAD(P)+) to drive metabolic transformations in and across most primary pathways. The eighth key metabolite is molecular oxygen (O2), thermodynamically activated for reduction by one electron path, leaving it kinetically stable to the vast majority of organic cellular metabolites.
Figures

References
-
- Jencks WP. Catalysis in chemistry and enzymology. Dover Publications; 1987.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
