

Different contributions of chemokine N-terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3
- PMID: 29272550
- PMCID: PMC5900987
- DOI: 10.1111/bph.14132
Different contributions of chemokine N-terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3
Abstract
Background and purpose: Chemokines and their receptors form an intricate interaction and signalling network that plays critical roles in various physiological and pathological cellular processes. The high promiscuity and apparent redundancy of this network makes probing individual chemokine/receptor interactions and functional effects, as well as targeting individual receptor axes for therapeutic applications, challenging. Despite poor sequence identity, the N-terminal regions of chemokines, which play a key role in their activity and selectivity, contain several conserved features. Thus far little is known regarding the molecular basis of their interactions with typical and atypical chemokine receptors or the conservation of their contributions across chemokine-receptor pairs.
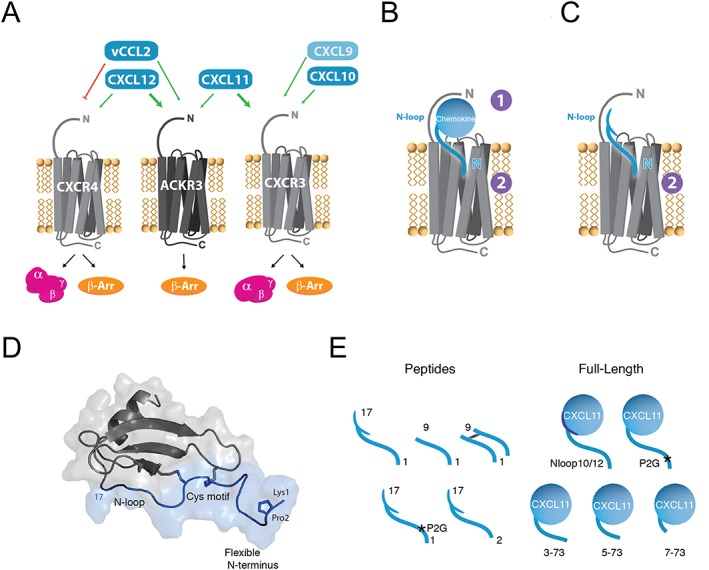
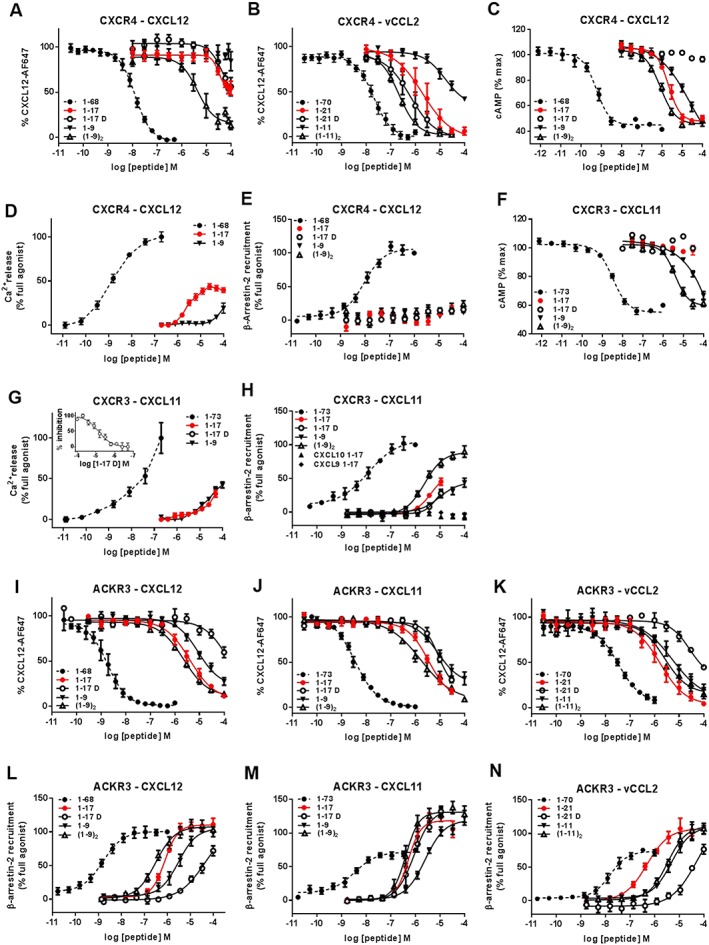
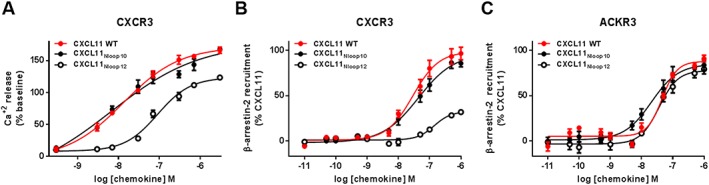
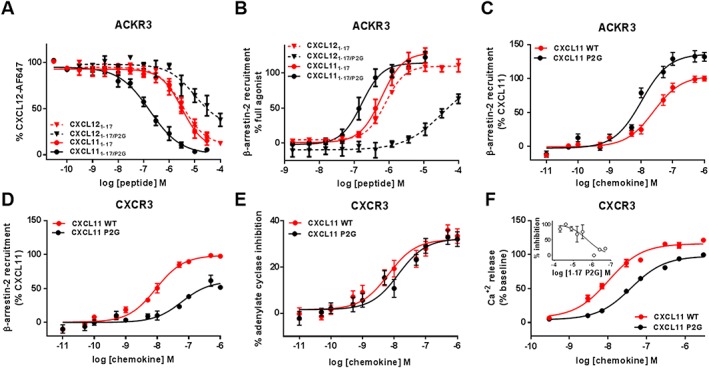
Experimental approach: We used a broad panel of chemokine variants and modified peptides derived from the N-terminal region of chemokines CXCL12, CXCL11 and vCCL2, to compare the contributions of various features to binding and activation of their shared receptors, the two typical, canonical G protein-signalling receptors, CXCR4 and CXCR3, as well as the atypical scavenger receptor CXCR7/ACKR3, which shows exclusively arrestin-dependent activity.
Key results: We provide molecular insights into the plasticity of the ligand-binding pockets of these receptors, their chemokine binding modes and their activation mechanisms. Although the chemokine N-terminal region is a critical determinant, neither the most proximal residues nor the N-loop are essential for binding and activation of ACKR3, as distinct from binding and activation of CXCR4 and CXCR3.
Conclusion and implications: These results suggest a different interaction mechanism between this atypical receptor and its ligands and illustrate its strong propensity to activation.
© 2017 The British Pharmacological Society.
Figures
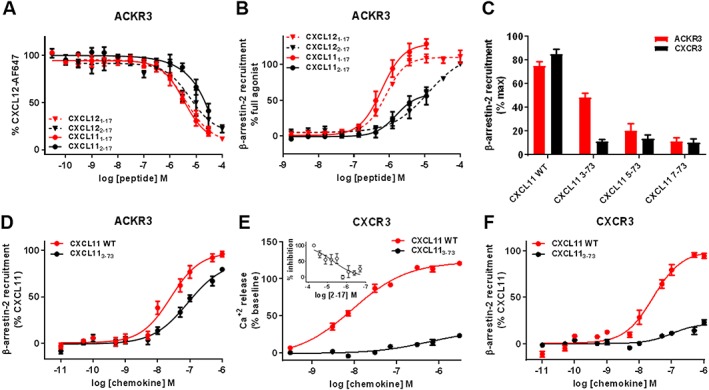
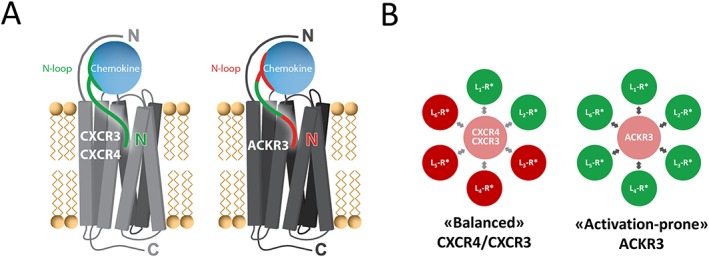
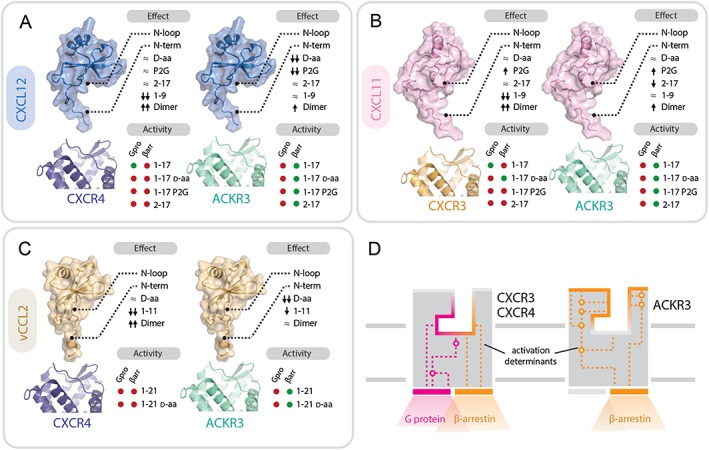
References
-
- Allen SJ, Crown SE, Handel TM (2007). Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25: 787–820. - PubMed
-
- Bachelerie F, Ben‐Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ et al (2014a). International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 66: 1–79. - PMC - PubMed
-
- Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R et al (2014b). New nomenclature for atypical chemokine receptors. Nat Immunol 15: 207–208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
