

Strain profiling and epidemiology of bacterial species from metagenomic sequencing
- PMID: 29273717
- PMCID: PMC5741664
- DOI: 10.1038/s41467-017-02209-5
Strain profiling and epidemiology of bacterial species from metagenomic sequencing
Abstract
Microbial communities are often composed by complex mixtures of multiple strains of the same species, characterized by a wide genomic and phenotypic variability. Computational methods able to identify, quantify and classify the different strains present in a sample are essential to fully exploit the potential of metagenomic sequencing in microbial ecology, with applications that range from the epidemiology of infectious diseases to the characterization of the dynamics of microbial colonization. Here we present a computational approach that uses the available genomic data to reconstruct complex strain profiles from metagenomic sequencing, quantifying the abundances of the different strains and cataloging them according to the population structure of the species. We validate the method on synthetic data sets and apply it to the characterization of the strain distribution of several important bacterial species in real samples, showing how its application provides novel insights on the structure and complexity of the microbiota.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
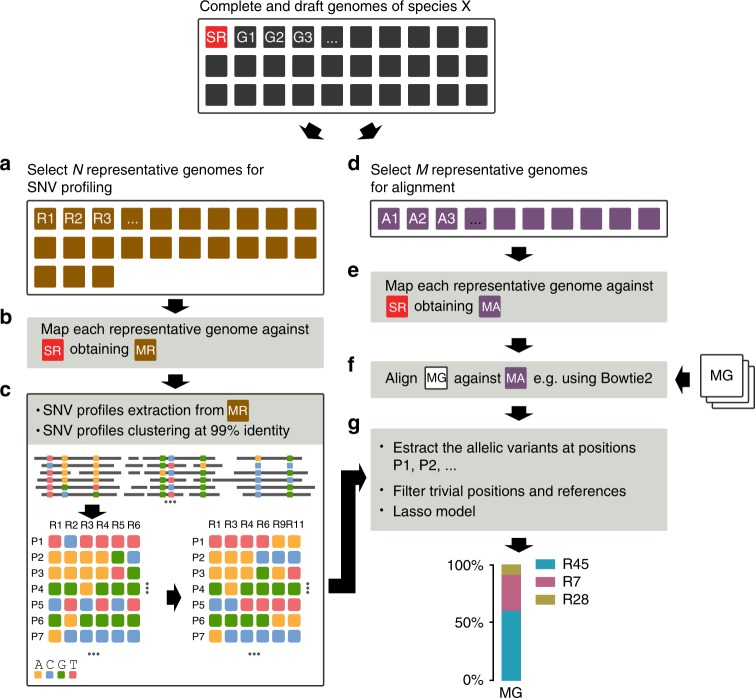
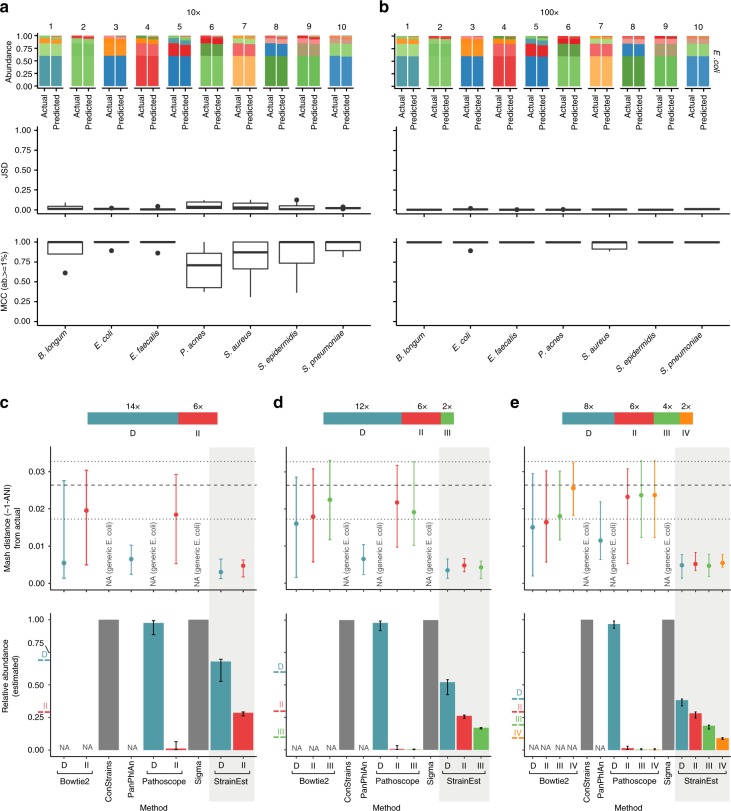
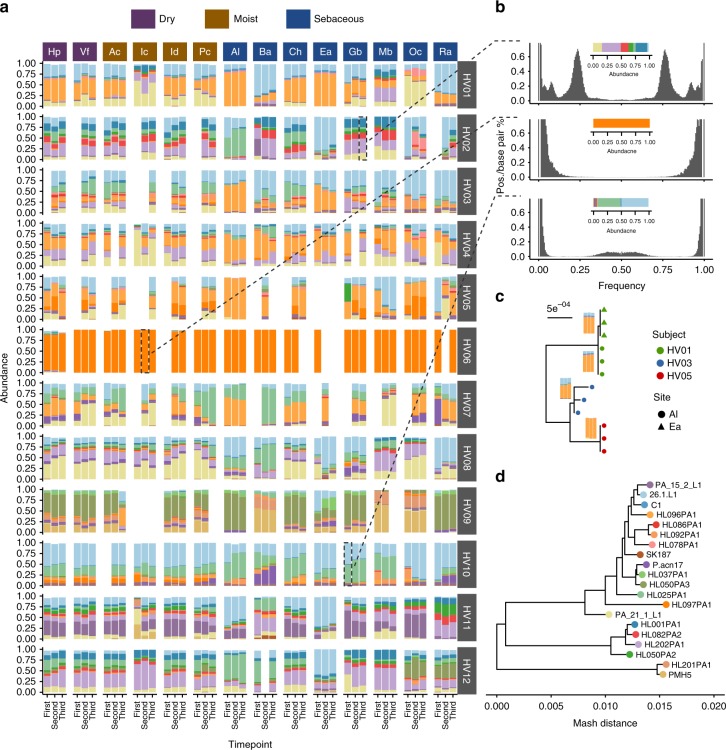
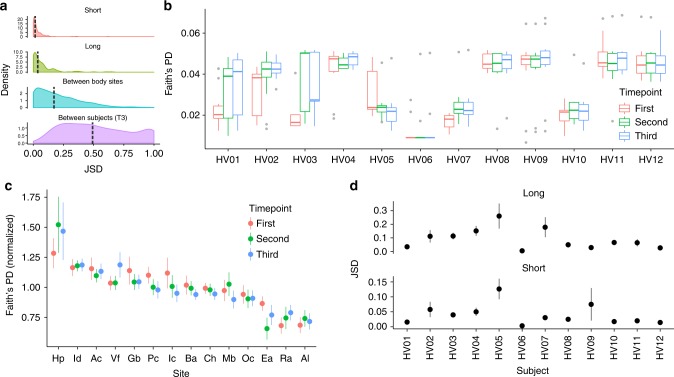
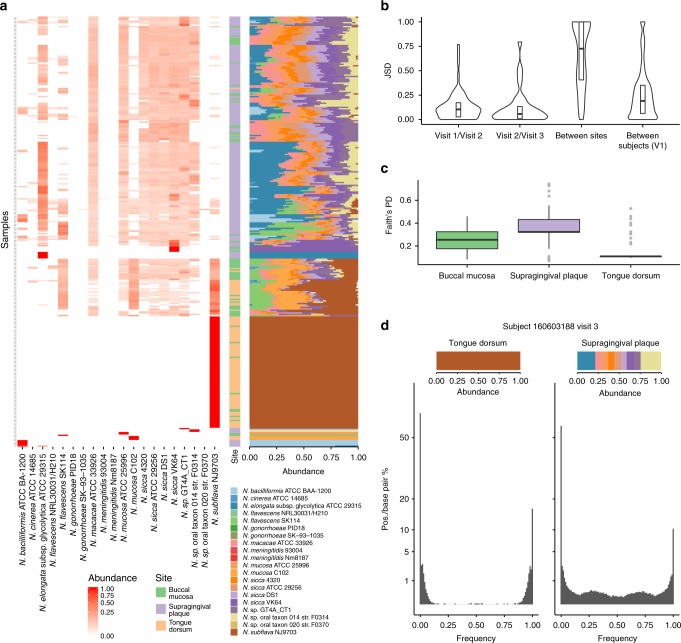
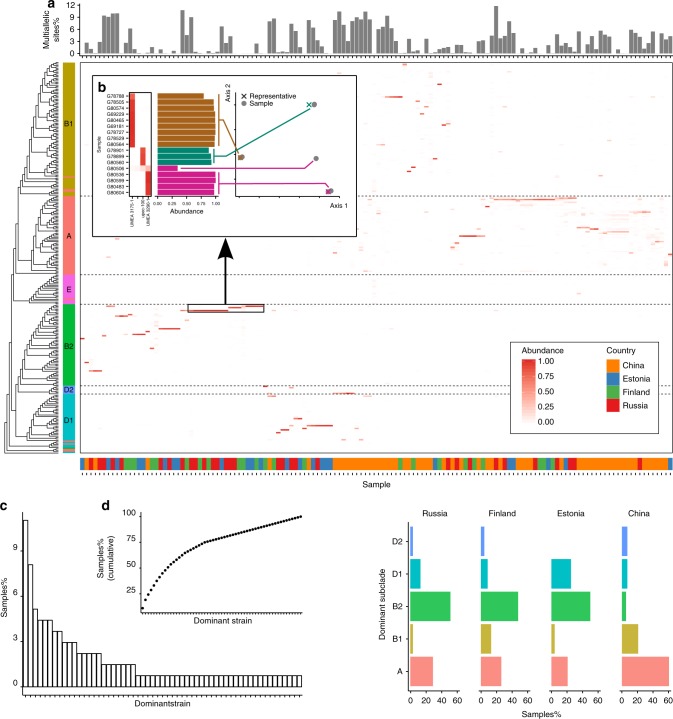
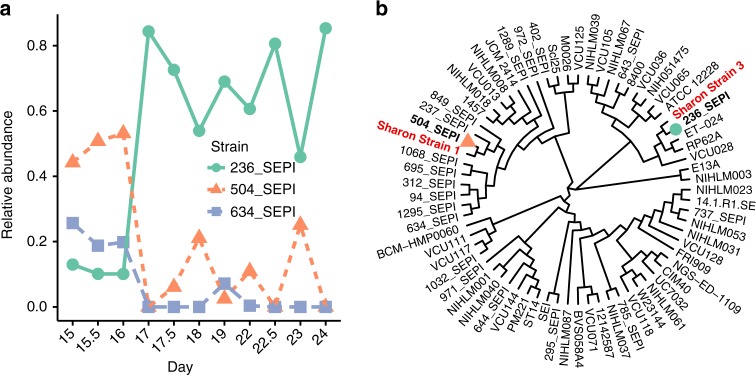
References
-
- Medini, D. et al. Microbiology in the post-genomic era. Nat. Rev. Microbiol.6, 419–430 (2008). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
