

A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes
- PMID: 29275994
- PMCID: PMC5764092
- DOI: 10.1016/j.devcel.2017.11.020
A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes
Abstract
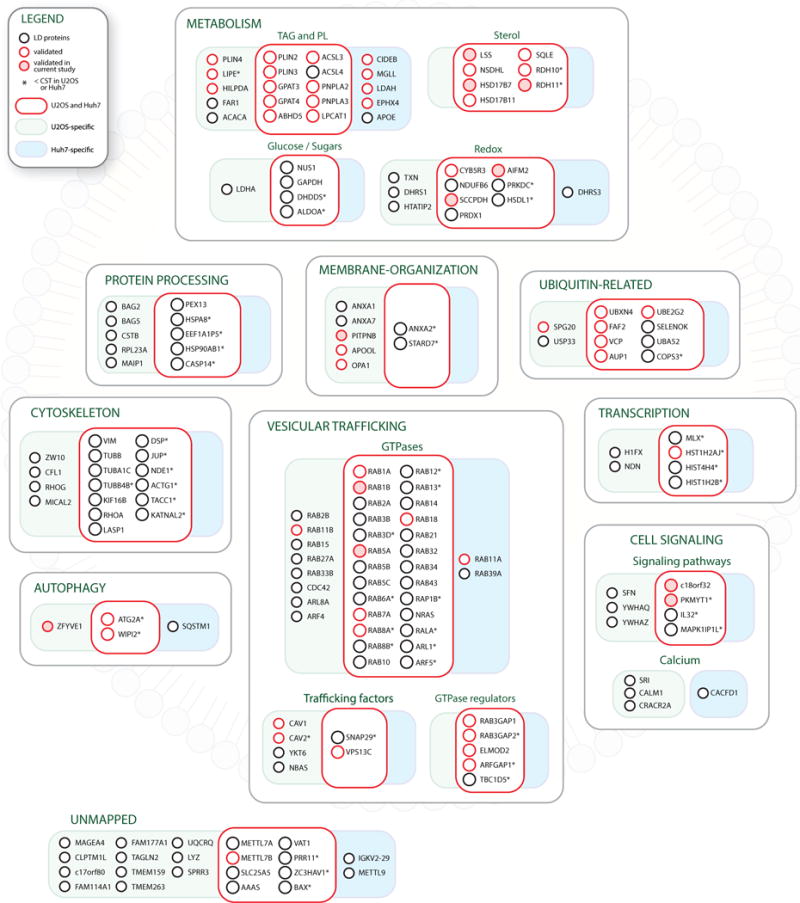
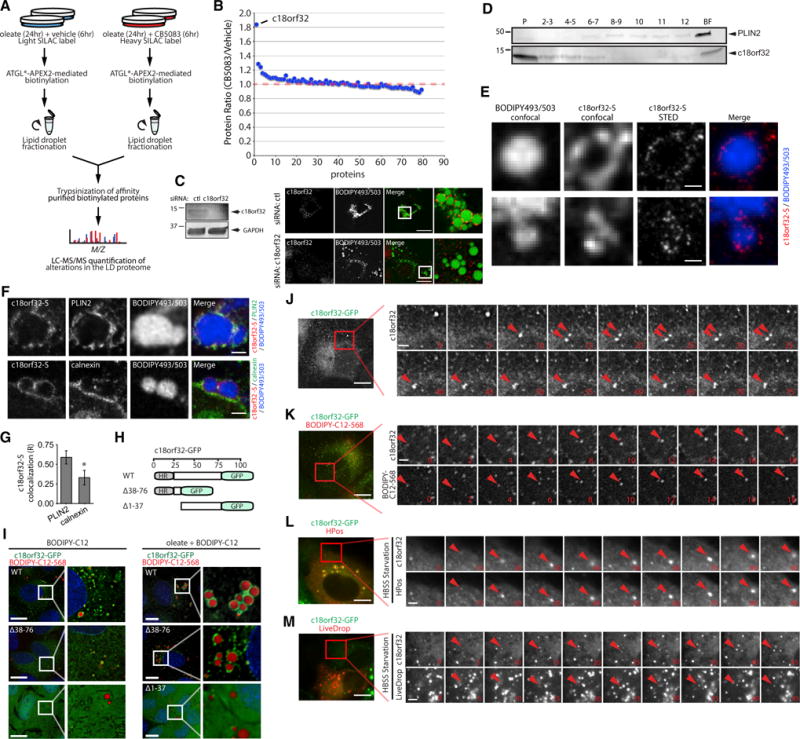
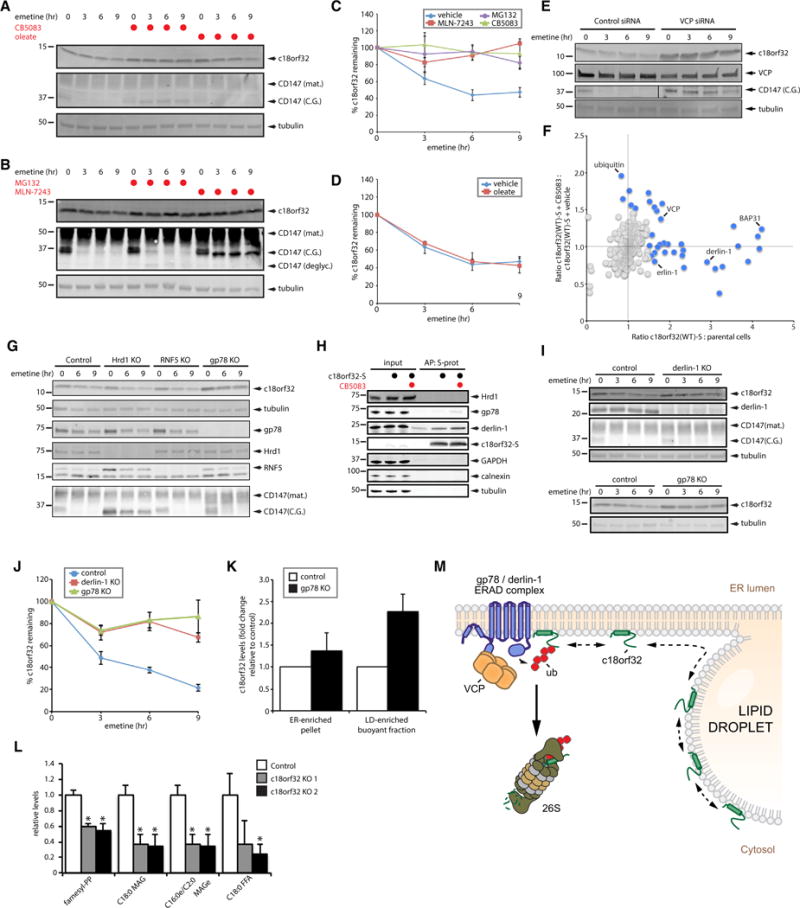
Lipid droplet (LD) functions are regulated by a complement of integral and peripheral proteins that associate with the bounding LD phospholipid monolayer. Defining the composition of the LD proteome has remained a challenge due to the presence of contaminating proteins in LD-enriched buoyant fractions. To overcome this limitation, we developed a proximity labeling strategy that exploits LD-targeted APEX2 to biotinylate LD proteins in living cells. Application of this approach to two different cell types identified the vast majority of previously validated LD proteins, excluded common contaminating proteins, and revealed new LD proteins. Moreover, quantitative analysis of LD proteome dynamics uncovered a role for endoplasmic reticulum-associated degradation in controlling the composition of the LD proteome. These data provide an important resource for future LD studies and demonstrate the utility of proximity labeling to study the regulation of LD proteomes.
Keywords: APEX; APEX2; ERAD; biotinylation; endoplasmic reticulum; lipid droplet; proteasome; proteome; proximity labeling; ubiquitin.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
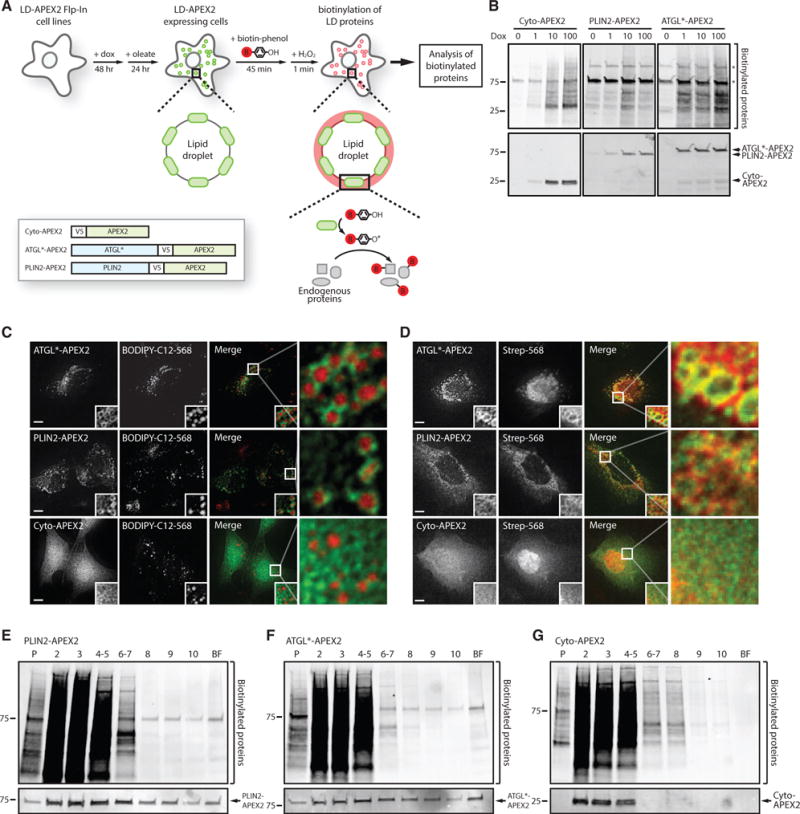
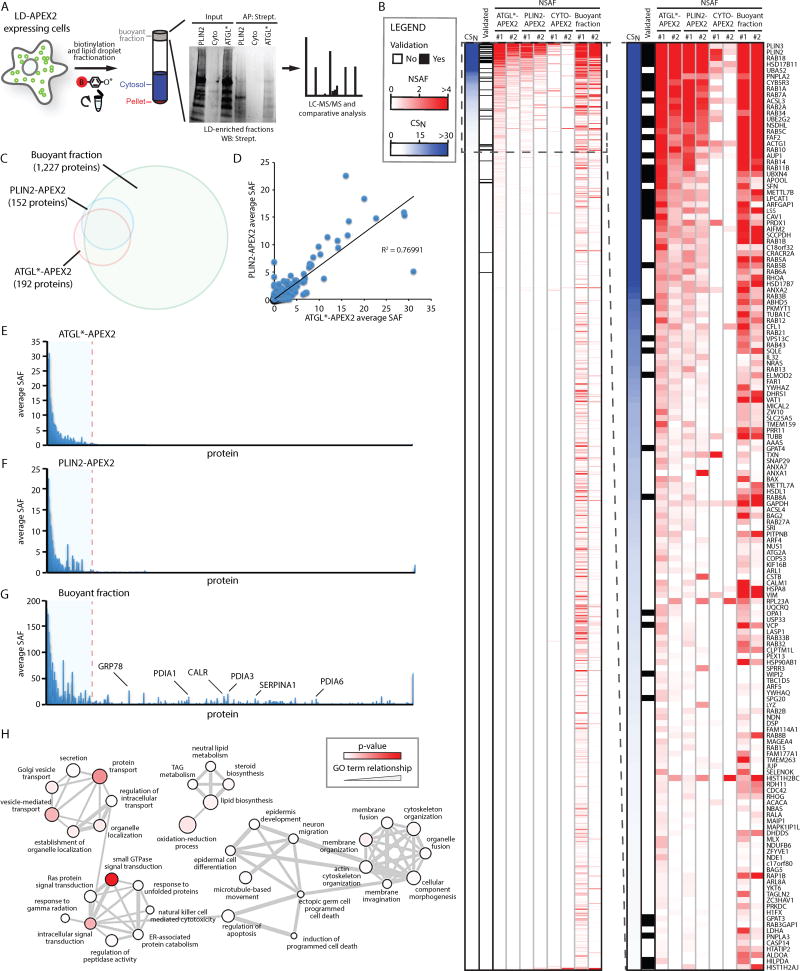
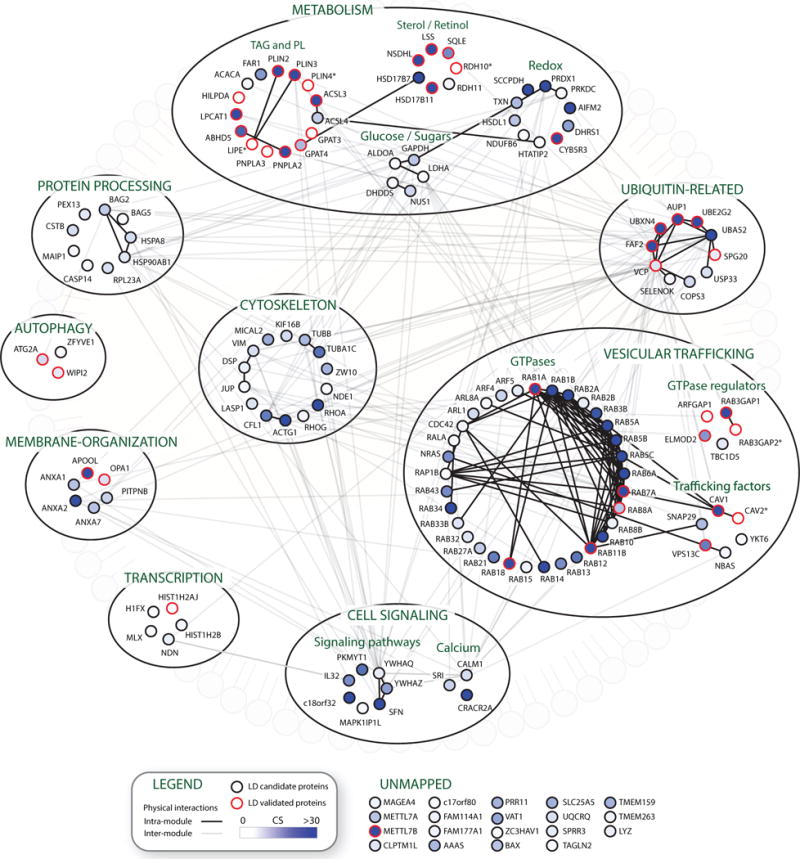
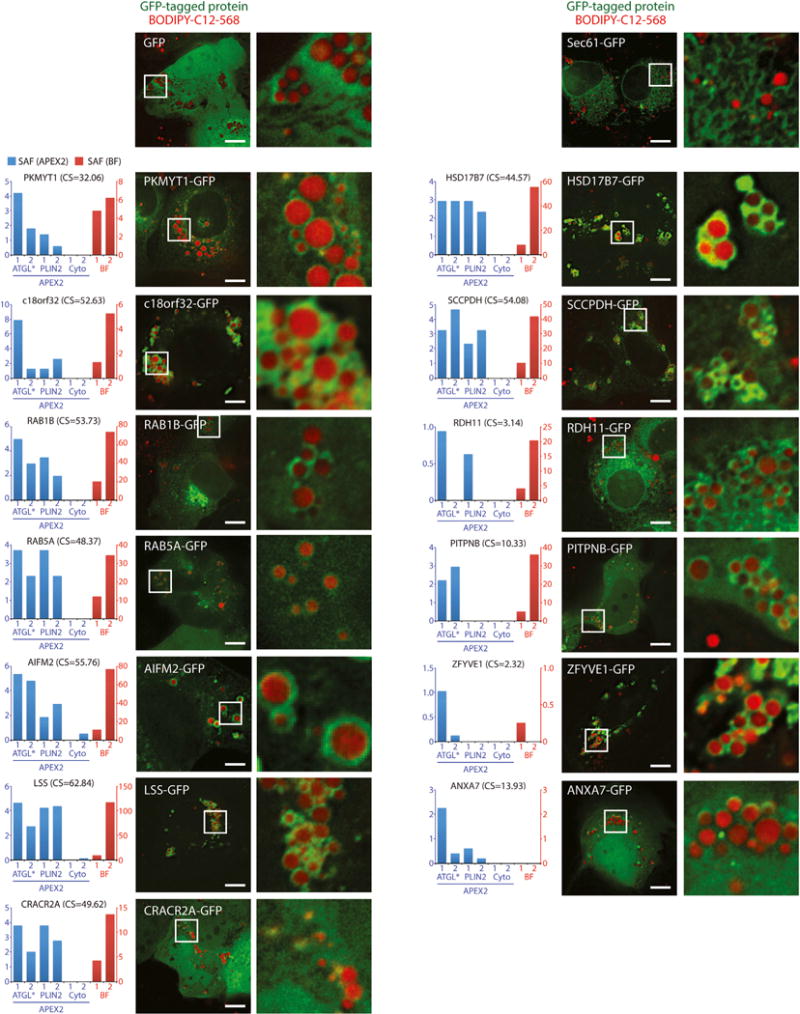
Comment in
-
Understanding the Lipid Droplet Proteome and Protein Targeting.Dev Cell. 2018 Jan 8;44(1):1-2. doi: 10.1016/j.devcel.2017.12.017. Epub 2018 Jan 8. Dev Cell. 2018. PMID: 29316437 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
